1 IMP中文版教程
IMP: bridging the gap for medicinal plant genomics
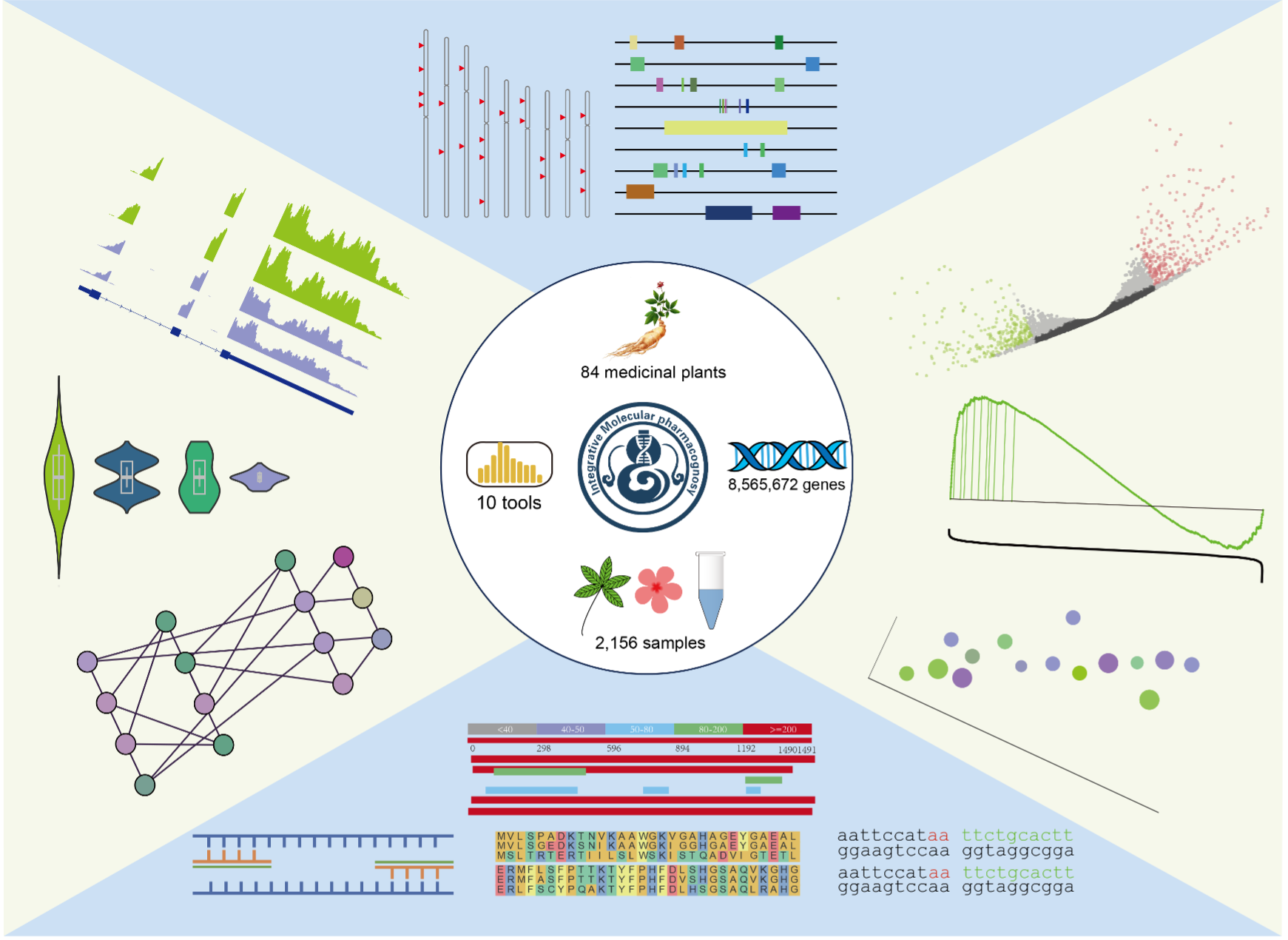
药用植物因其潜在的抗肿瘤、抗炎和抗氧化特性,在民族医学和传统医学中引起了极大的关注。基因组测序和合成生物学的最新进展重新激发了人们对这些天然产物的兴趣。尽管有很多药用植物的基因组和转录组测序数据,但缺乏可公开访问的基因注释和表格格式的基因表达数据,这不利于它们的有效利用。为了解决这一紧迫问题,我们开发了IMP (Integrated Medicinal Plantomics)整合药用植物组学平台(https://www.bic.ac.cn/IMP)。IMP收录了84个高质量的基因组(预期收录所有植物的基因组),整理了848,565,672个基因,以及2,158个转录组测序样本,涵盖了多个器官、组织、发育阶段和胁迫刺激。通过集成的10个分析模块,用户可以简单地在IMP中探索基因的注释、序列、功能、分布和表达。IMP的开发和使用将会从基础数据层面促进药用植物分子代谢途径的解析,进而在推动合成生物学的发展、促进药物发现和药物生产的天然来源的探索方面发挥重要作用。
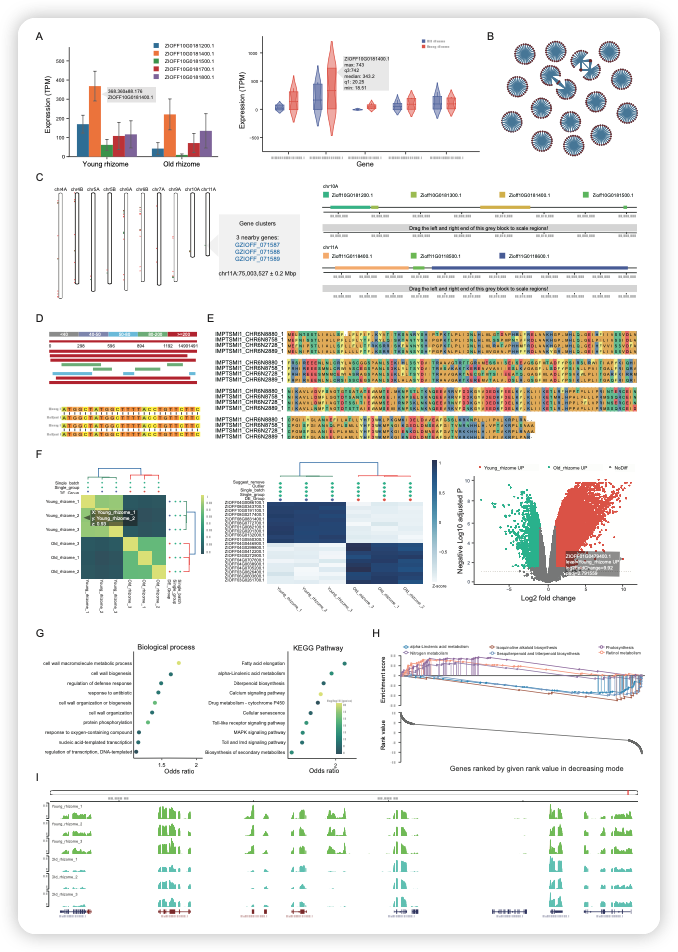
IMP 针对收录的数据提供了 10 个功能分析模块,示例性结果如下图,包括多基因表达图谱的绘制、共表达基因的搜寻和鉴定、基因簇的展示、BLAST 序列搜索、多序列比对、在线差异基因分析(样品相关性热图、差异基因热图和火山图)、GO/KEGG富集分析、GSEA 富集分析、IGV 基因组浏览器展示、引物设计、序列提取等。
 ## 文章发表 {#publication}
## 文章发表 {#publication}
IMP 于 2023 年 10 月发表于国际著名期刊 Nucleic Acids Research, https://doi.org/10.1093/nar/gkad898。
中国中医科学院中药资源中心陈同博士和杨莓博士(在读)为论文第一作者,黄璐琦院士、郭娟(国家万人计划)、袁媛(杰青)为该论文共同通讯作者。相关工作得到中央级公益性科研院所基本科研业务费专项资金、中国中医科学院科技创新工程项目、道地药材品质保障与资源持续利用全国重点实验室开放课题、国家重点研发专项等的支持。
陈同,博士,中国中医科学院副教授 研究方向涉及高通量数据分析、生物信息工具开发、合成生物学、表观组学等,在Cell Stem Cell (封面文章),Nucleic Acids Research,Nature communications, Protein & Cell, iMeta等高水平杂志以第一或通讯作者发表文章十余篇;开发在线绘图和分析平台 ImageGP、BIC、EVenn, 在线数据库平台IMP、CRAMdb、ETCM1/2、CMNPD、iLABdb、SoFDA 等,总计使用超 150 万人次;运营有十四万人关注的微信公众号和视频号《生信宝典》,分享有1400 多篇生物信息分析原创文章、教程和视频,阅读播放千万次。联合创办iMeta期刊,现为执行主编,致力于打造微生物和生物信息领域的国产高水平杂志。
杨莓,天津中医药大学,中药学在读博士生。目前研究方向为中药基因组,转录组等数据分析和多组学工具开发。以第一作者或共同第一作者在Nucleic Acids Research,Frontiers in Plant Science,Phytochemical Analysis杂志发表论文3篇,副主编著作1部,参编著作1部。
黄璐琦,中共党员,中国工程院院士。现任国家中医药管理局副局长、党组成员,中国中医科学院院长。任中国科协第十届全国委员会副主席,国家监察委员会第一届特约监察员,第十五届、十六届北京市人民代表大会代表。政协第十三届全国委员会常务委员。荣获全国优秀共产党员称号。从事中药资源学与分子生药学研究。提出和发展了“分子生药学”学科,揭示和阐明了道地药材形成理论,建立珍稀濒危常用中药资源五种保护模式,组织实施第四次全国中药资源普查,摸清我国中药资源本底。获国家科学技术进步二等奖5项、全国创新争先奖章、国家杰出青年科学基金资助、中国标准创新突出贡献奖、全国优秀博士学位论文指导教师等荣誉。主持研发我国首个具有完全自主知识产权的治疗新冠肺炎的中药新药,荣获全国抗击新冠肺炎疫情先进个人称号。以第一作者或通讯作者发表论文580余篇,包括BMJ,NAT COMMUN,PNAS,JACS等SCI论文300余篇。
袁媛,中国中医科学院首席研究员,博士生导师,国家杰出青年科学基金获得者。任中国中医科学院中药资源中心副主任,兼任中华中医药学会中药鉴定分会主任委员,中国中西医结合学会分子生药学专业委员会副主任委员,中国中药协会种子种苗专委会副主任委员,主要从事中药鉴定与分子生药学研究。先后获中国青年女科学家奖团队奖、北京市科技新星、科技部中青年科技创新领军人才、国家中医药管理局青年岐黄学者、中国科协求是杰出青年成果转化奖等。近5年获国家科技进步二等奖、中国中西医结合学会科技进步一等奖、中华中医药学会科技进步二等奖等7项,在Nature communications、Science Bulletin 等杂志上以第一或通讯作者发表论文120余篇,授权专利43项,发布国家标准、团体标准11项。
郭娟,2010年博士毕业于中国科学院武汉植物园,中国中医科学院中药资源中心研究员。入选国家级科技创新领军人才、青年岐黄学者、中国药学发展奖杰出青年学者、中华中医药学会中青年创新人才,获国家自然科学基金优秀青年基金资助。主要从事中药资源与分子生药学研究,在PNAS、Nature Communications等学术期刊共发表文章60余篇,授权专利10项,成果转化2项,兼任中华中医药学会青年委员会常务委员、中国植物学会药用植物与植物药专业委员会委员、中国中西医结合学会分子生药学专业委员会委员。主要研究方向:中药活性成分活性成分生物合成及调控、药用植物代谢工程研究、天然产物合成生物学、修饰酶结构与作用机制及其分子设计与进化。
引文: IMP: bridging the gap for medicinal plant genomics. Nucleic Acids Research, gkad898, https://doi.org/10.1093/nar/gkad898
1.1 首页信息
数据平台访问地址https://www.bic.ac.cn/IMP/。首页采用平面组合布局,分为导航、网站描述、统计信息和功能展示 4 个部分。
- Logo 设计体现药物特色,
药葫芦+DNA双螺旋体现药用植物分子信息,辅以祥云标识,展示中国特色; - 轮播图和文字描述网站特色;首页的搜索为
全局检索,用户输入基因名字、通路信息或任意基因功能相关单词即可搜索目标基因,开启网站的探索之旅; - 右侧 2 个视频图标可以跳转当前页面可用功能的具体描述:国内跳转 B 站,国外跳转 YouTube 平台。
- 中间 4 个图标列出数据库收录数据的统计信息:基因数目、基因碱基数、样品数和物种数目。
- 下面 12 个模式图列出网站的主要功能和功能跳转。
- 最后是网站的更新日志(最近新增的模块,功能还在完善中)。

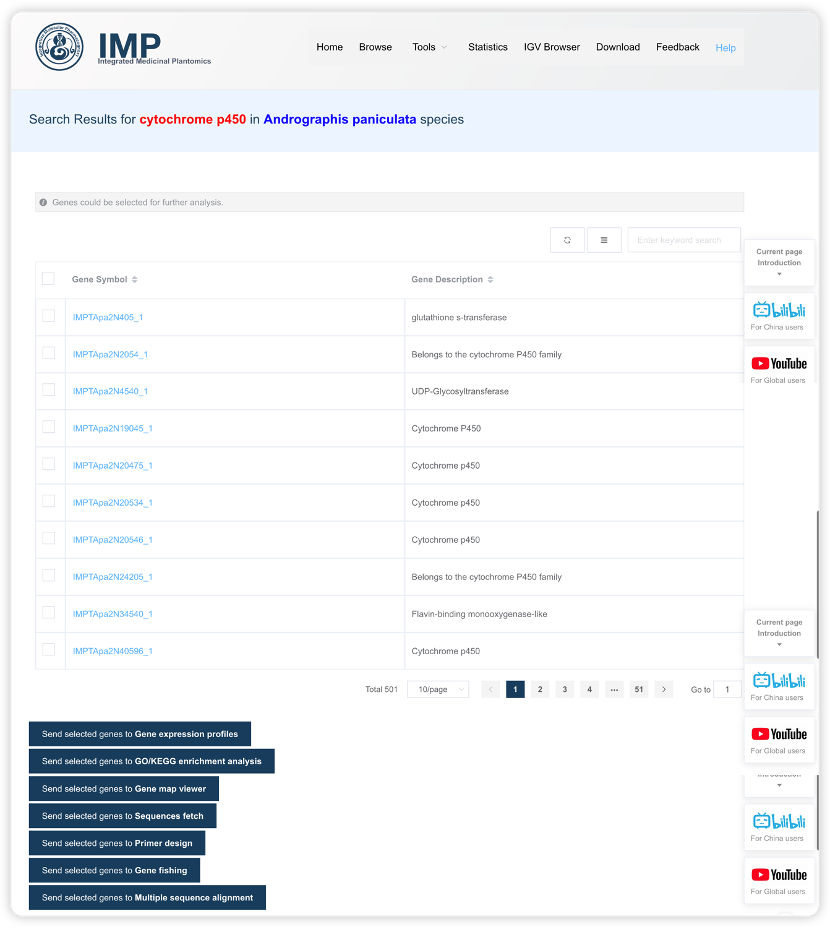
1.2 基于功能描述、注释或基因名字的全局搜索
在首页的全局搜索框中输入基因的名字、基因的功能描述或基因的 GO 注释/KEGG通路注释的信息,即输入任何文字都可以去匹配出关注的基因(当然也有一些文字什么都匹配不出来)。比如默认选中的物种是穿心莲,默认输入的文字是cytochrome p450,我们需要做的就是点击Submit 提交一下,新标签页会出现搜索结果。
如果碰到页面不出来的情况下,请看下浏览器最上部菜单栏下面是否有窗口被拦截的提示。
搜索结果页面的标识条,会用红字标记搜索的文字信息, 蓝字标记选择的物种信息。下面的表格列出所有的搜索结果,分页展示:
- 可以选择一页展示的条目数增减搜索结果的数目,也可以选择展示所有条目。
- 可以在右上角搜索框进行二次检索,进一步聚焦要关注的基因。
- 右上角也可以调节表格中展示哪些列,默认只有 2 列信息,可加列。
独特的
Send to快捷操作很多物种的基因名字都是 ID 类似的编号,通常记不住。IMP 可以通过文字或序列的方式搜索出一系列相关基因,选择后,点击
Send to就可以把这些目标基因集发送到对应的功能模块,实现免输入 Gene ID的快速操作。比如查看搜索出的 CYP450 的整体表达信息、基因组的分布信息、批量序列提取、引物设计和多序列比对等。
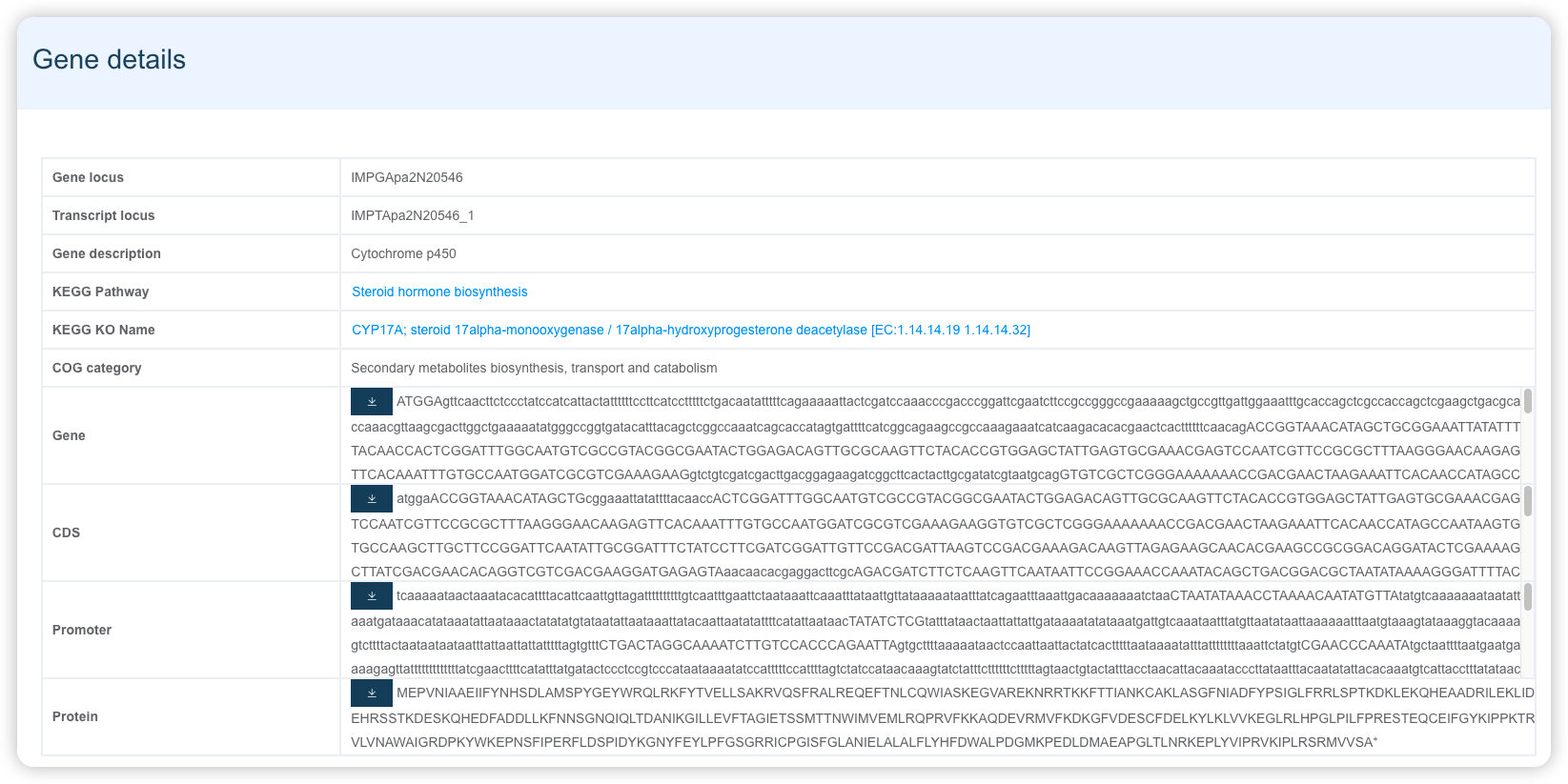
1.3 以单基因为中心的详情页面展示
页面分为 3 个部分:
- 第一部分展示基因的基本信息,包括名字信息、功能描述信息和序列信息。
- 第二部分展示基因在不同数据集的表达图谱信息。
- 用户可以选择数据的预处理方式、图形的布局、箱体的排序、数据集来调整展示的内容。
- 同时可以通过
padding调整图的左、下、右的空间,以免发生文字溢出。 - 最终的截图图可导出
SVG格式,用于文章组图。 - SVG 图也可以在 BIChttps://www.bic.ac.cn/BIC/ 的 SVGEdit 平台进行简单编辑 http://www.ehbio.com/SVGEdit/editor/。

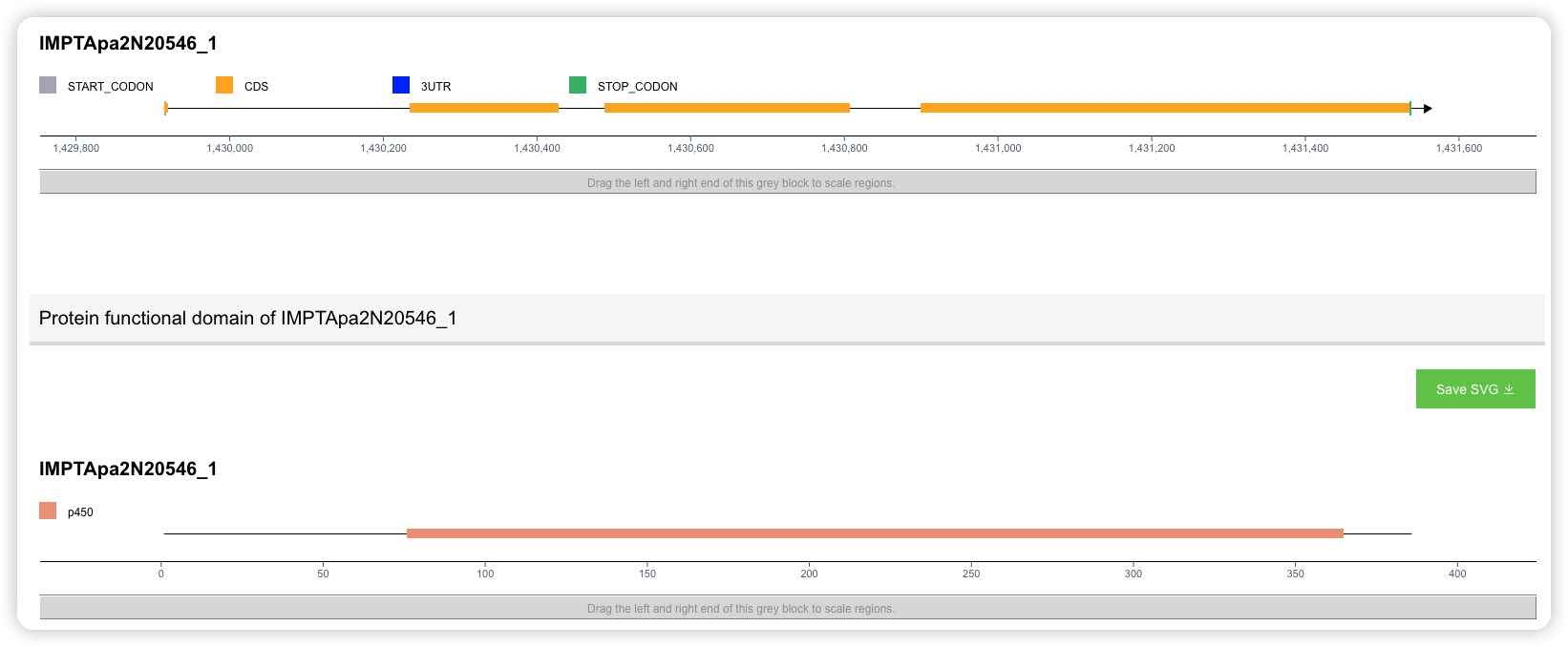
- 第三部分展示基因的结构(内含子、外显子、UTR 等信息)和蛋白功能域信息。
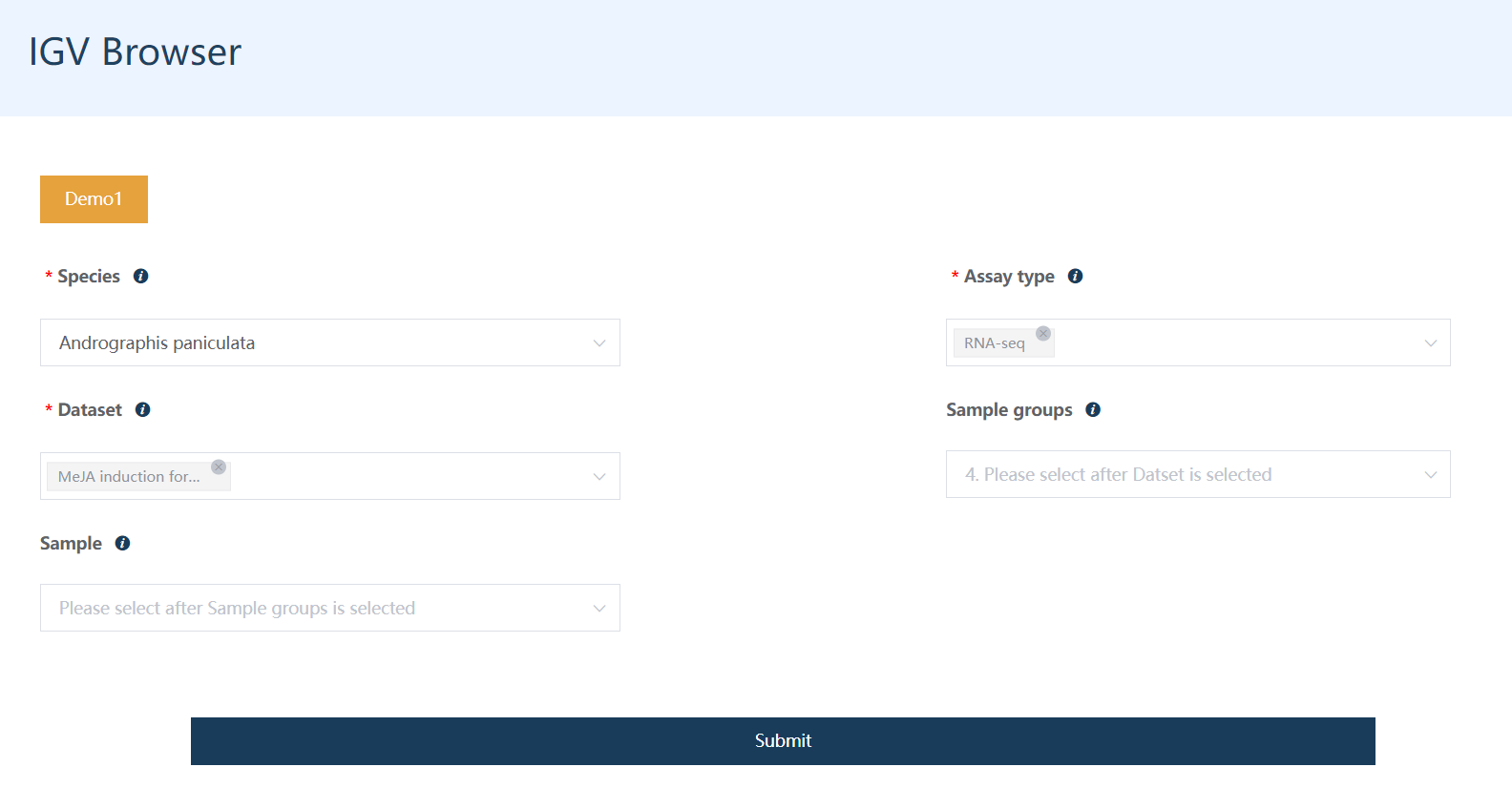
1.4 多基因表达图谱
可以自己按页面选择物种、数据集、样品(非必选的选项如果不选,默认是全选)、输入基因,也可以从搜索结果中直接带过来基因列表。
模糊搜索:支持根据基因的功能描述关键词进行模糊搜索,获取基因名,用于研究一类基因的表达图谱。
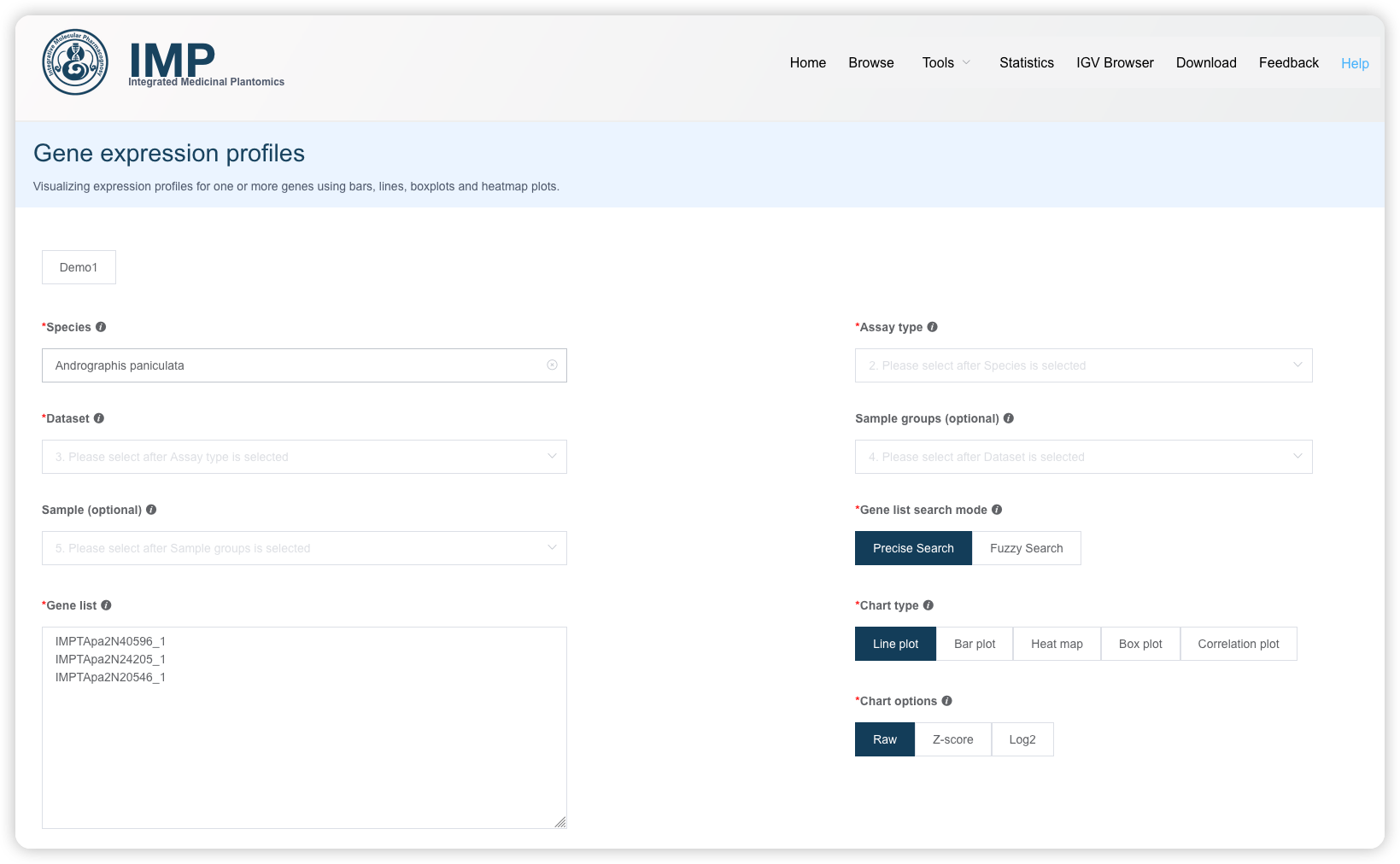
提交后获得基因表达图谱展示。
- 用户可以跳转图形的 padding 信息和高度信息
- 可视化结果可以导出 SVG 格式
- 作图数据可以下载,导入 ImageGP/BIC平台进行再次分析

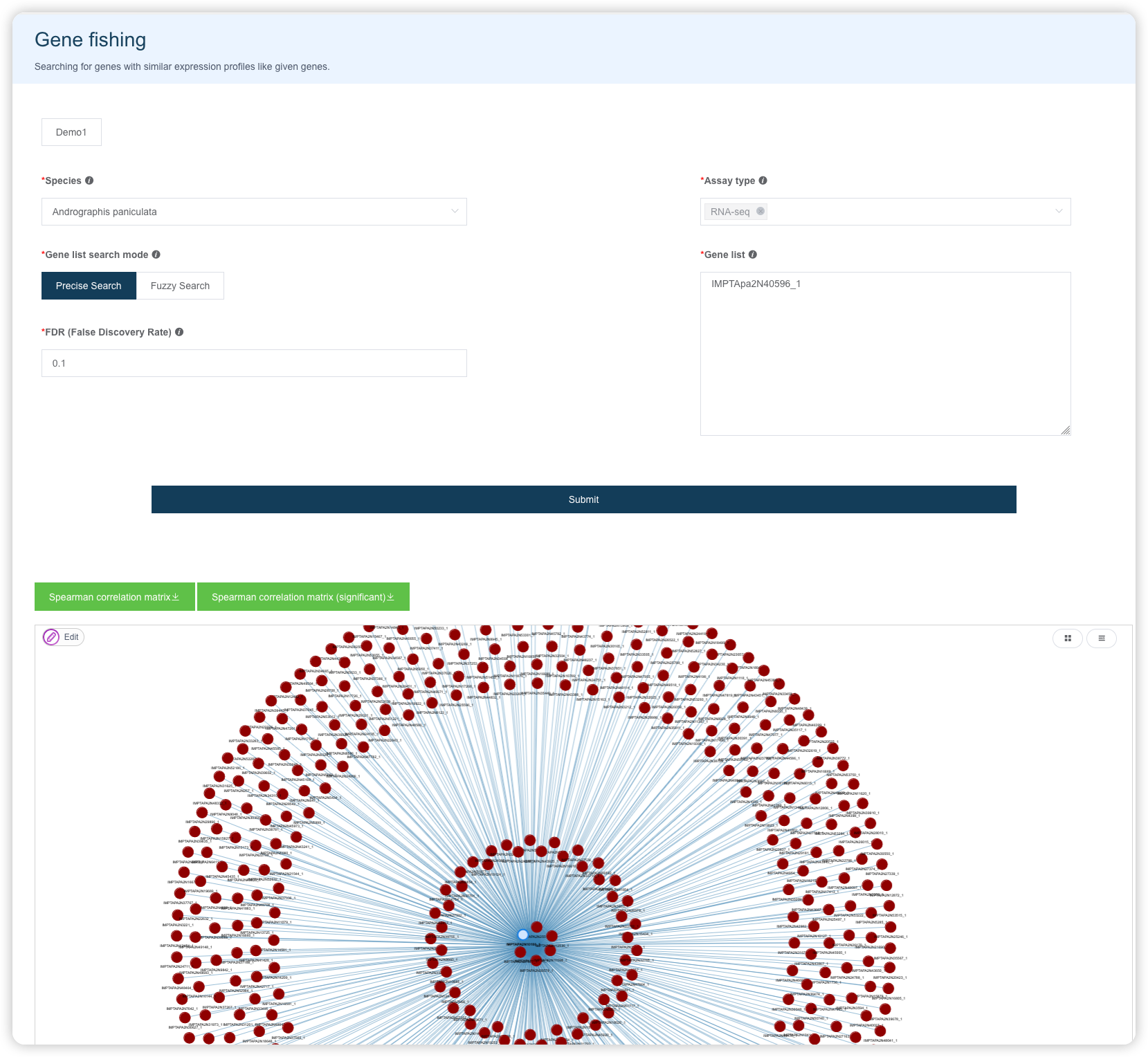
1.5 Gene fishing 调取表达模式相近的基因
选择物种、Assay type、匹配模式,输入基因名(可以通过Send to功能从其它页面发送过来),提交后获得一个相关性网络图和对应的结果数据。
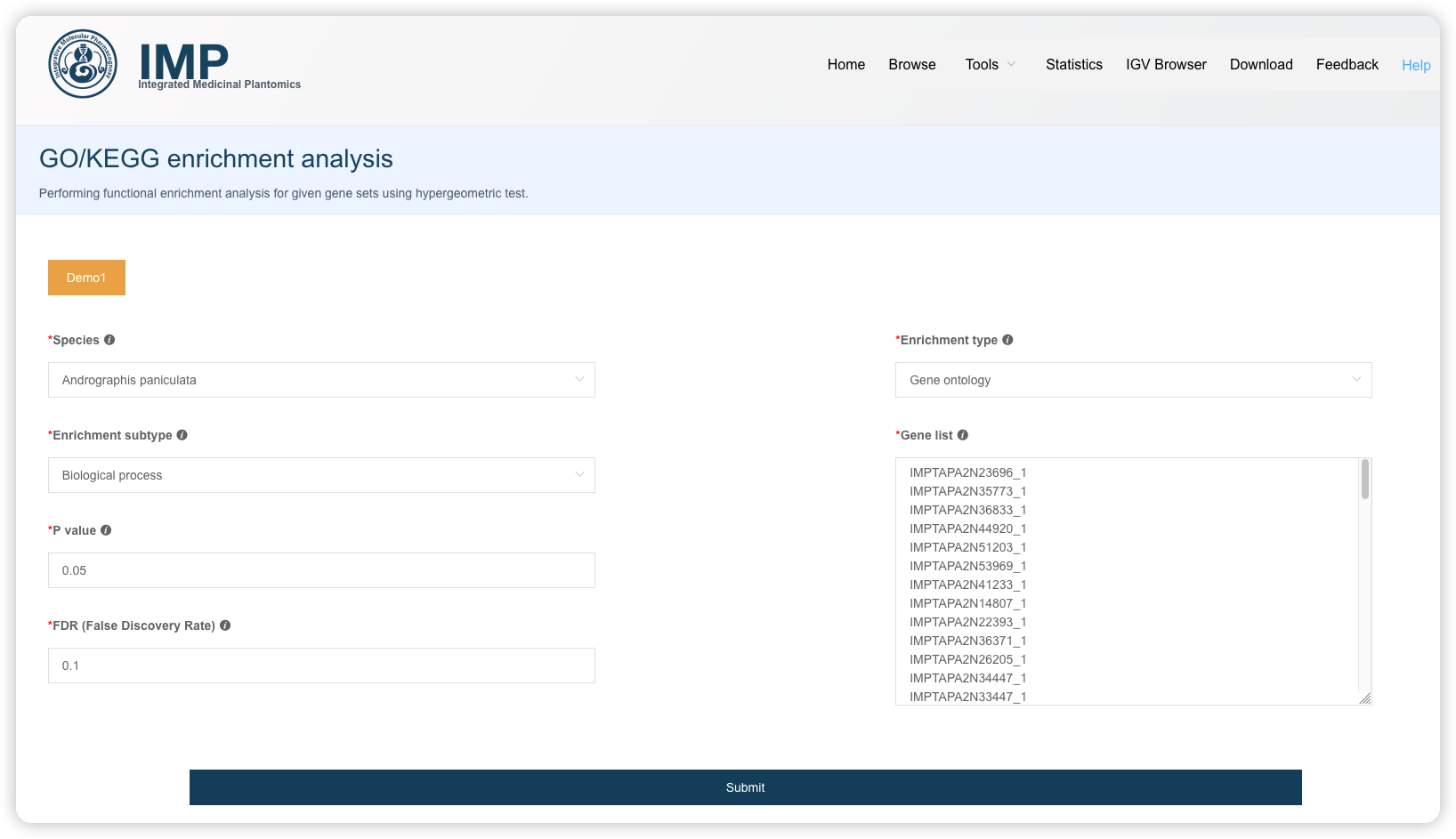
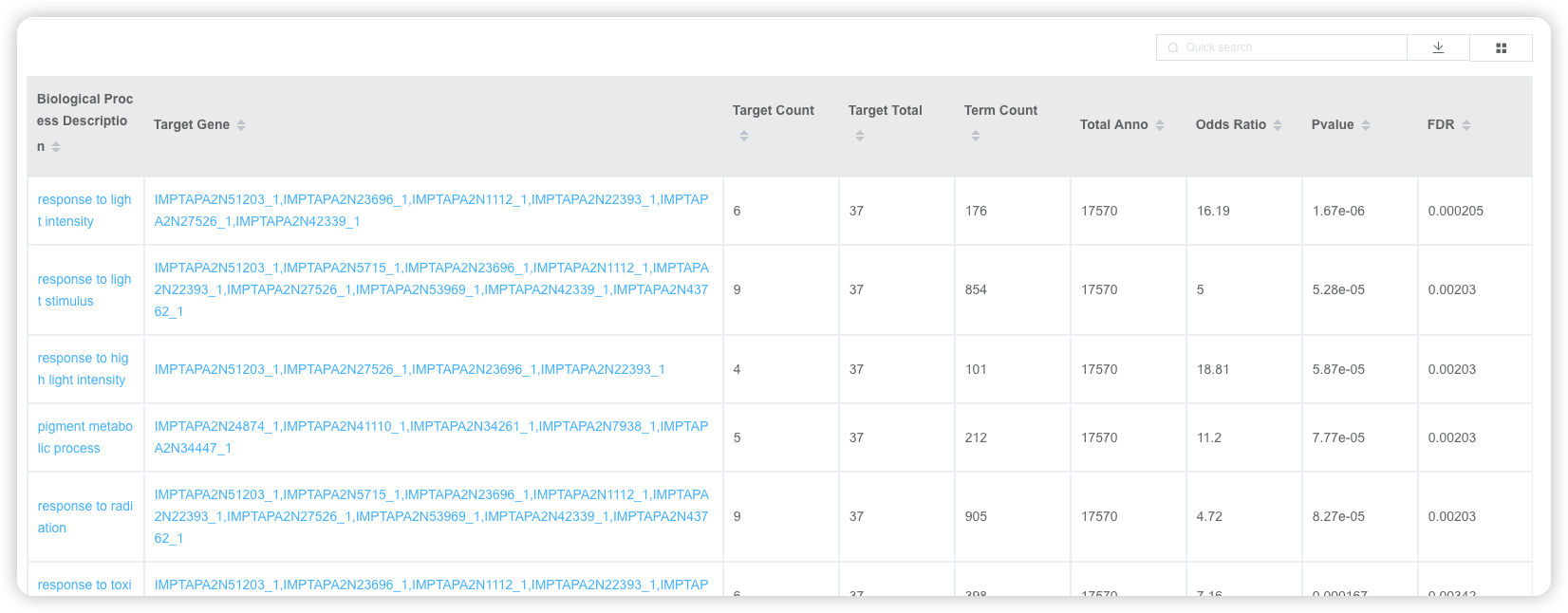
1.6 GO/KEGG 富集分析
用户选择物种,输入基因名字,即可进行GO/KEGG富集分析。阅读推文https://mp.weixin.qq.com/s/BCB16M4yI5Qa1tKyZy7WMg或查看视频https://www.bilibili.com/video/BV1rD4y1272a?p=4了解 GO/KEGG 富集分析的基本原理。
 点击后,可调整富集分析结果的配色方案、选择富集的条目进行展示。也可以下载表格文件,到高颜值免费在线绘图平台 ImageGP/BIC https://www.bic.ac.cn/BIC进行自由绘制。
点击后,可调整富集分析结果的配色方案、选择富集的条目进行展示。也可以下载表格文件,到高颜值免费在线绘图平台 ImageGP/BIC https://www.bic.ac.cn/BIC进行自由绘制。
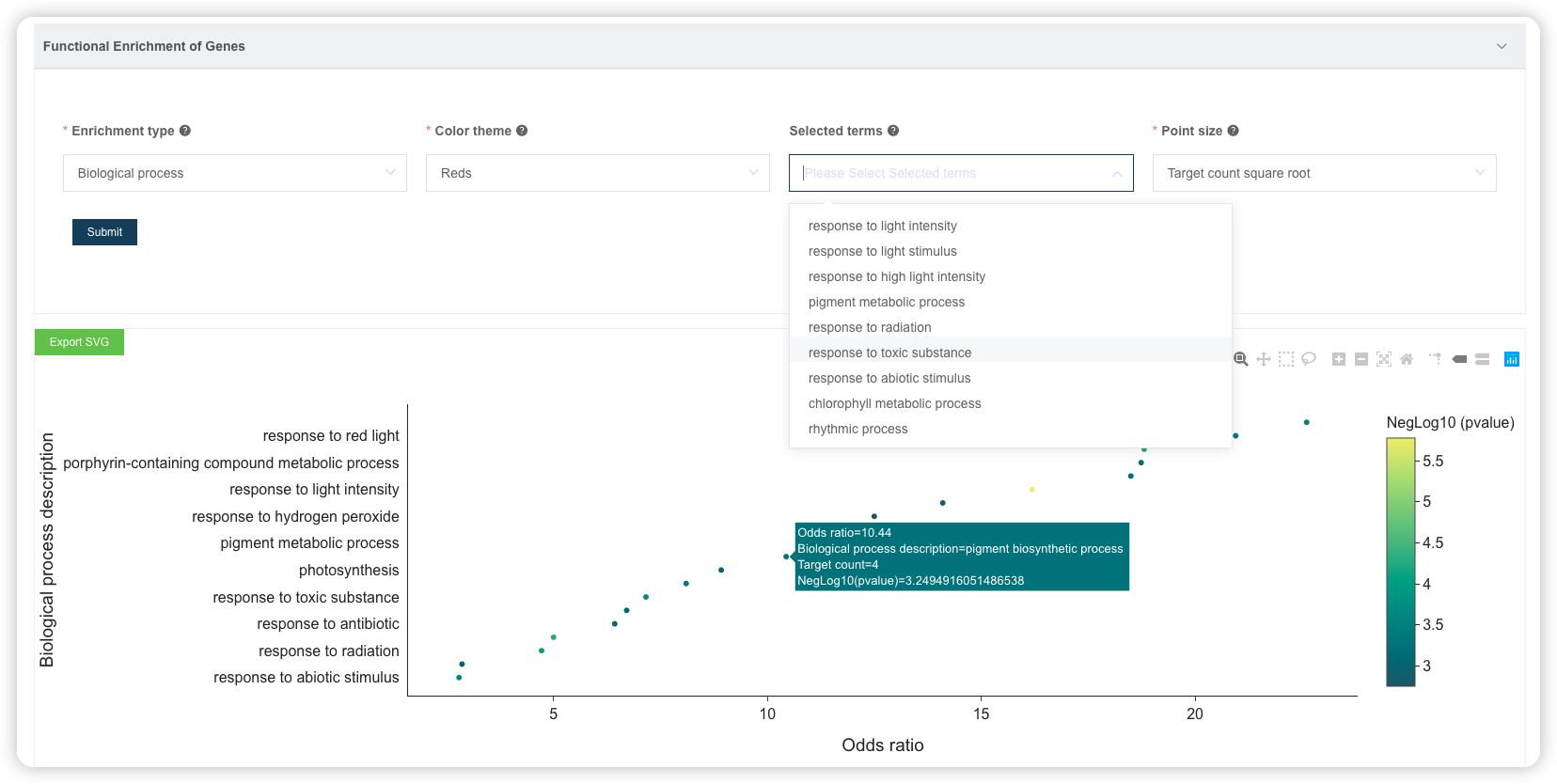
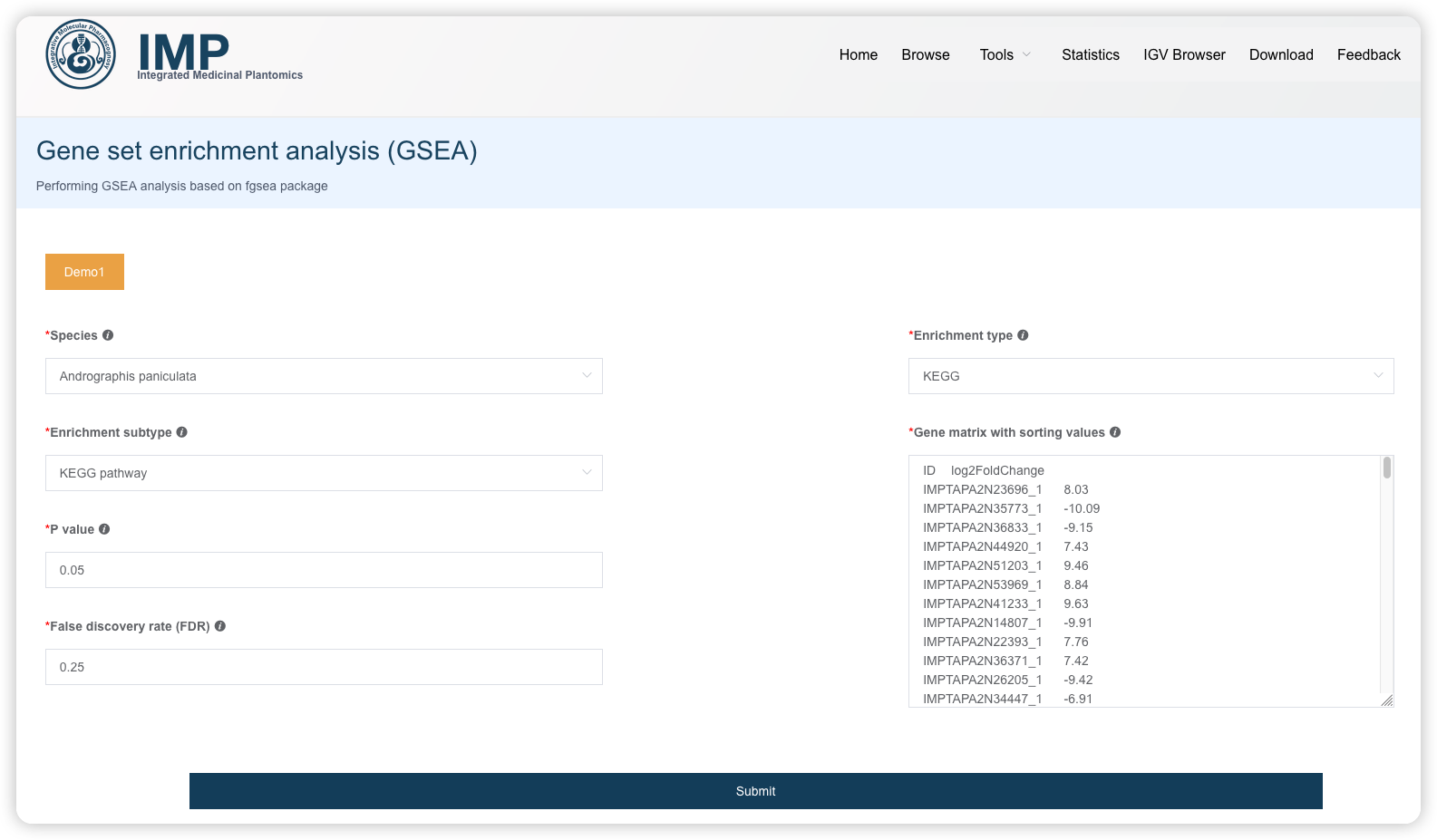
1.7 GSEA 富集分析
GSEA 富集分析的输入会麻烦一些,目前只支持包含一列基因和一列排序值的 2 列矩阵格式;排序值可以是常见的log2(fold change),p-value或也可以是其他定量值。
阅读推文https://mp.weixin.qq.com/s/WiYUUALSmb9v5gYVxmjwjA或查看视频https://www.bilibili.com/video/BV1rD4y1272a?p=5了解 GSEA富集分析的输入数据、原理和结果解读。
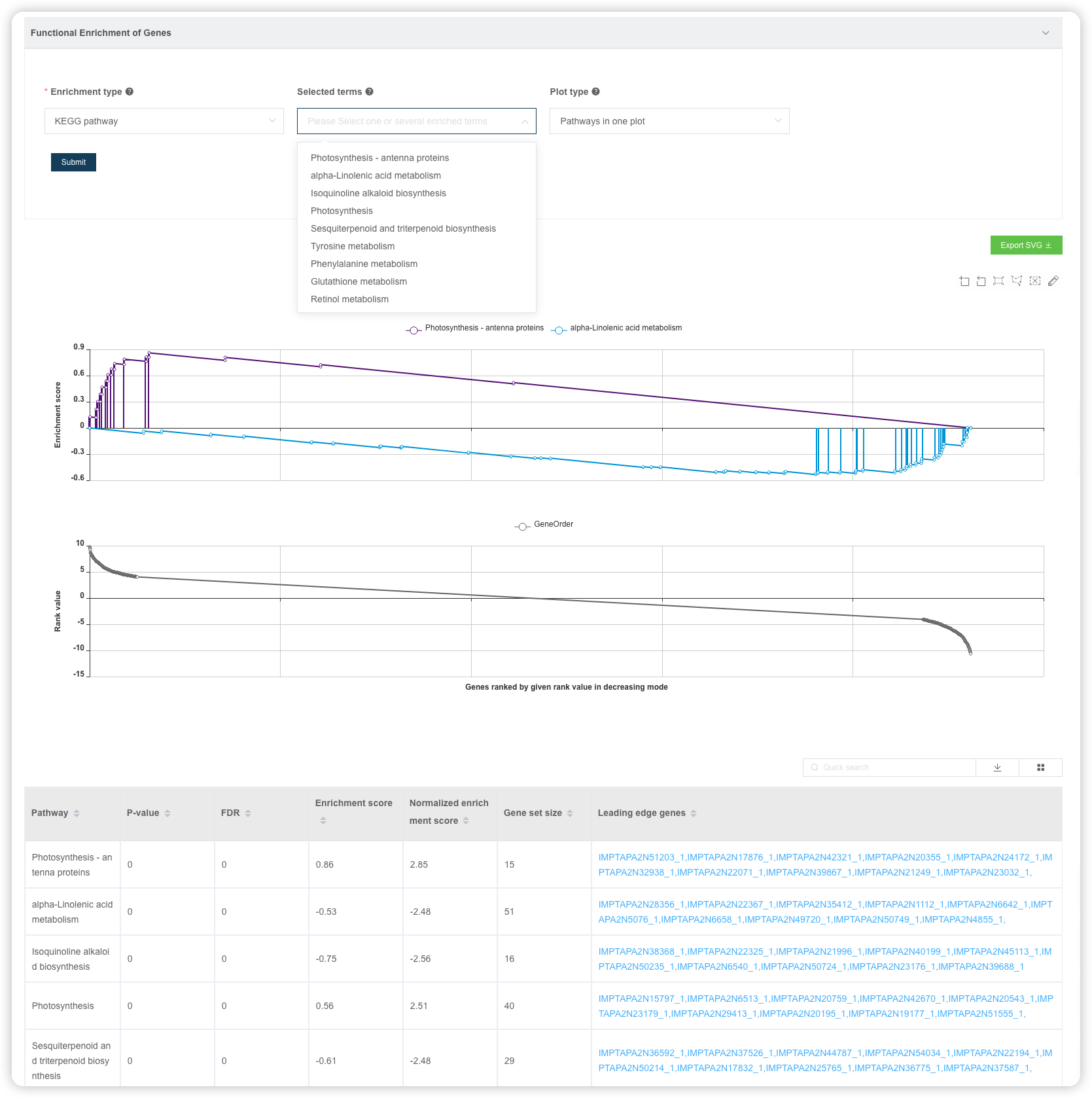
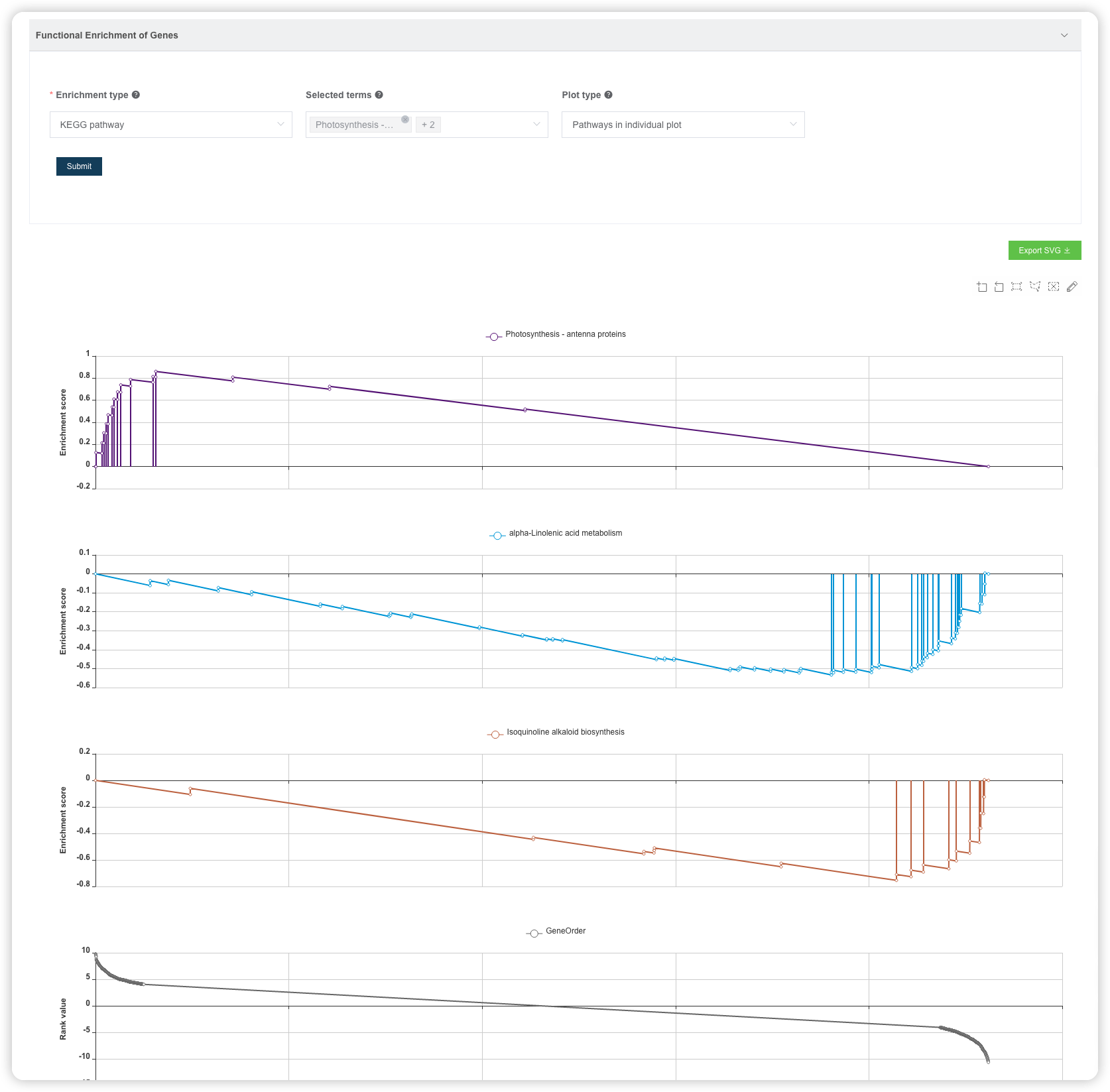
默认绘制最富集的 2 条通路在一张图上,可以自己选择绘制哪些通路,也可以将通路绘制在多张图上。
1.8 BLAST序列比对和搜索
BLAST 是鼎鼎有名的序列搜索工具,这里支持
- BLASTN: nucleotide to nucleotide
- TBLASTN: protein to translated nucleotide
- BLASTP: protein to protein
- BLASTX: translated nucleotide to protein
非模式物种常常没有统一的Gene Symbol,使用的是各种意义不明的 ID,序列搜索是把文献或私藏的序列映射到 IMP 或在 IMP 中搜索序列相似基因的好方法。这就是 BLAST 功能所做的。
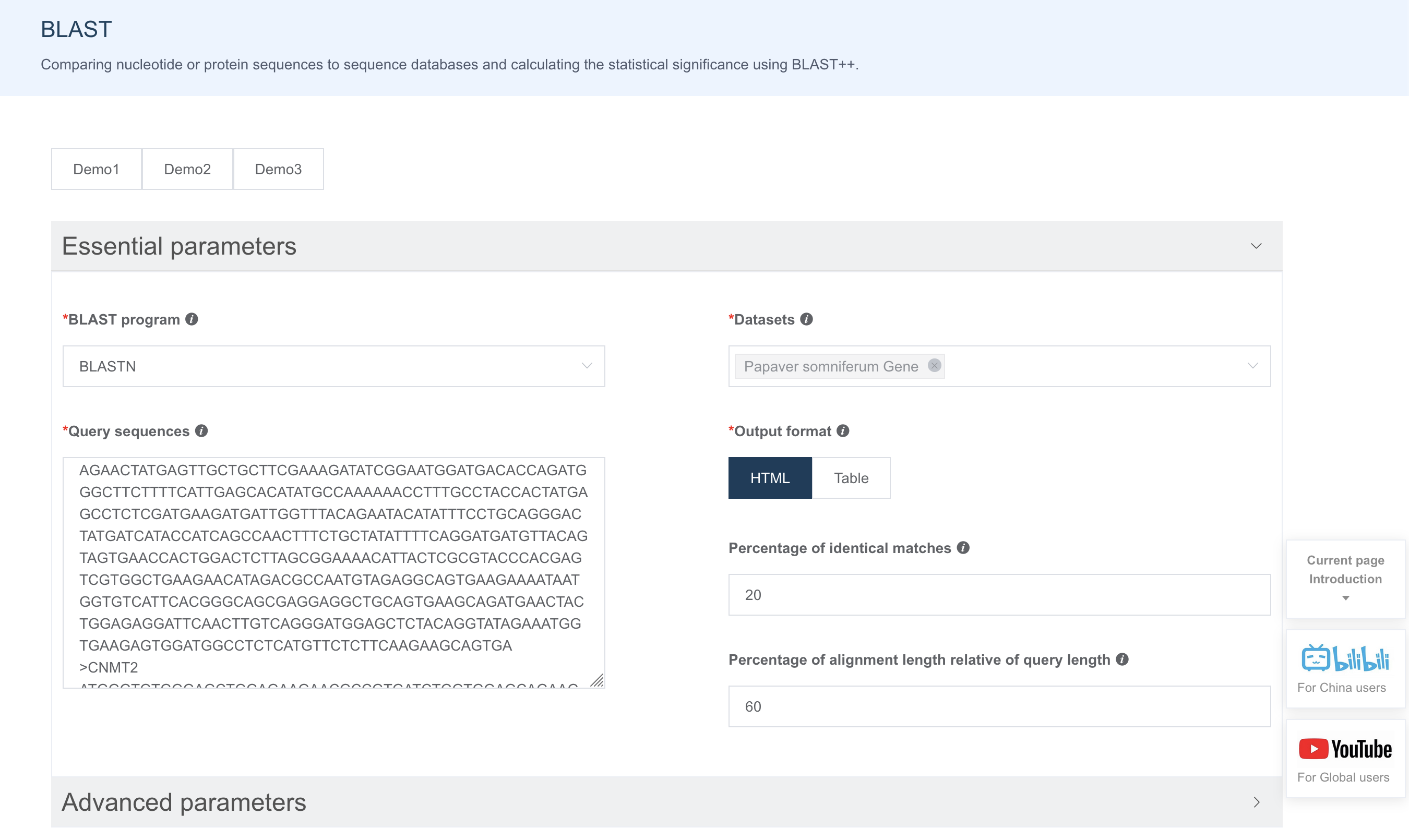
IMP 的 Blast 功能支持用户输入单条或多条 FASTA 序列进行搜索,用户也可以选择一个或多个或全部数据集。Advanced parameter处可以设置更多匹配控制参数。
HTML格式的输出会包含匹配区域的序列比对信息。如果用户输入了多条查询序列,可在Results for后面的下拉框中进行选择切换。
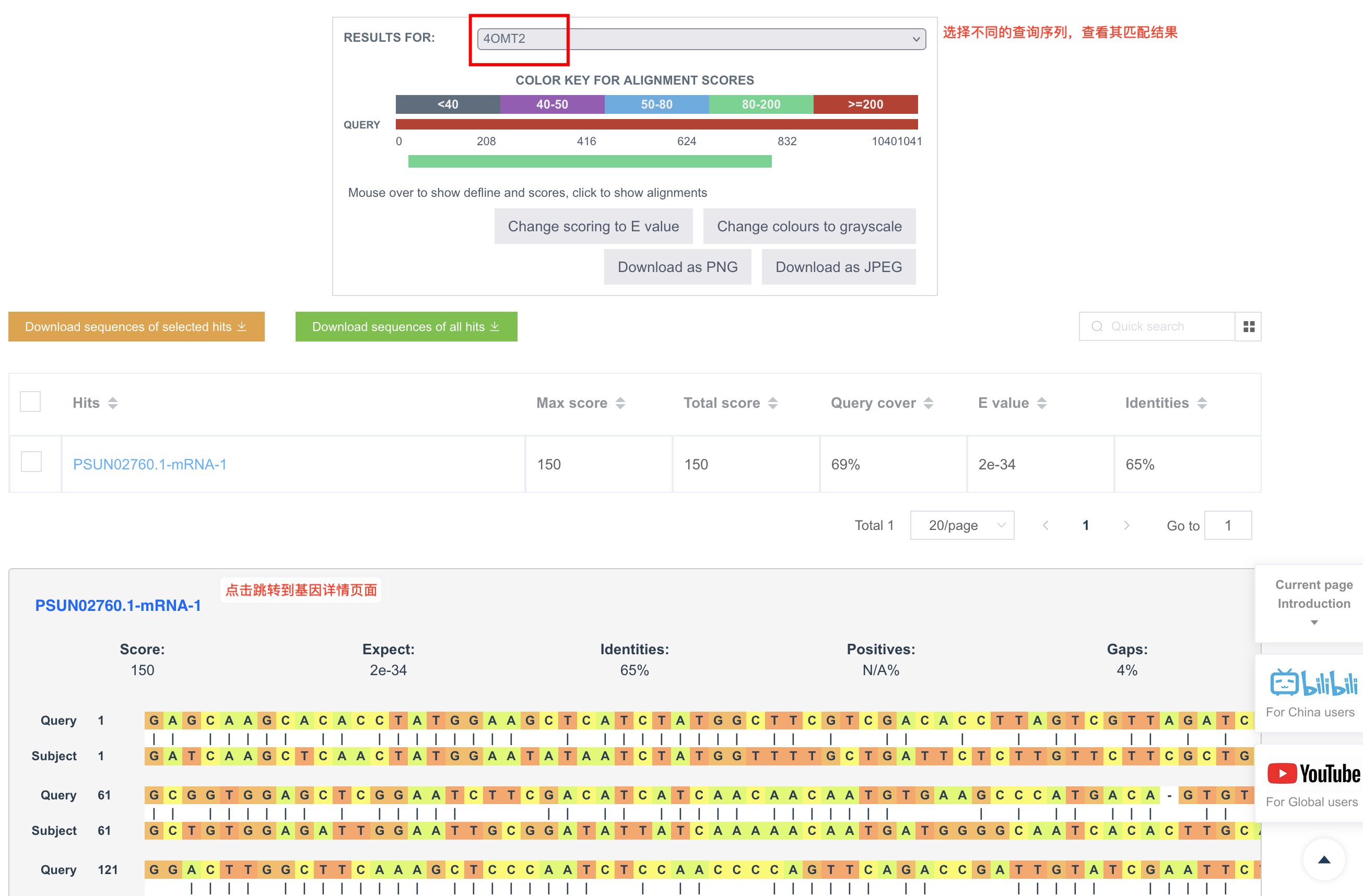
Table格式简洁明确地列出每条查询序列在数据库中的匹配序列,可以把匹配出的序列通过Send to功能发送到更多工具页面,快捷使用。
因为 BLAST自身的问题,如果用户选了多个数据库文件,当前会强制输出 Table 格式。
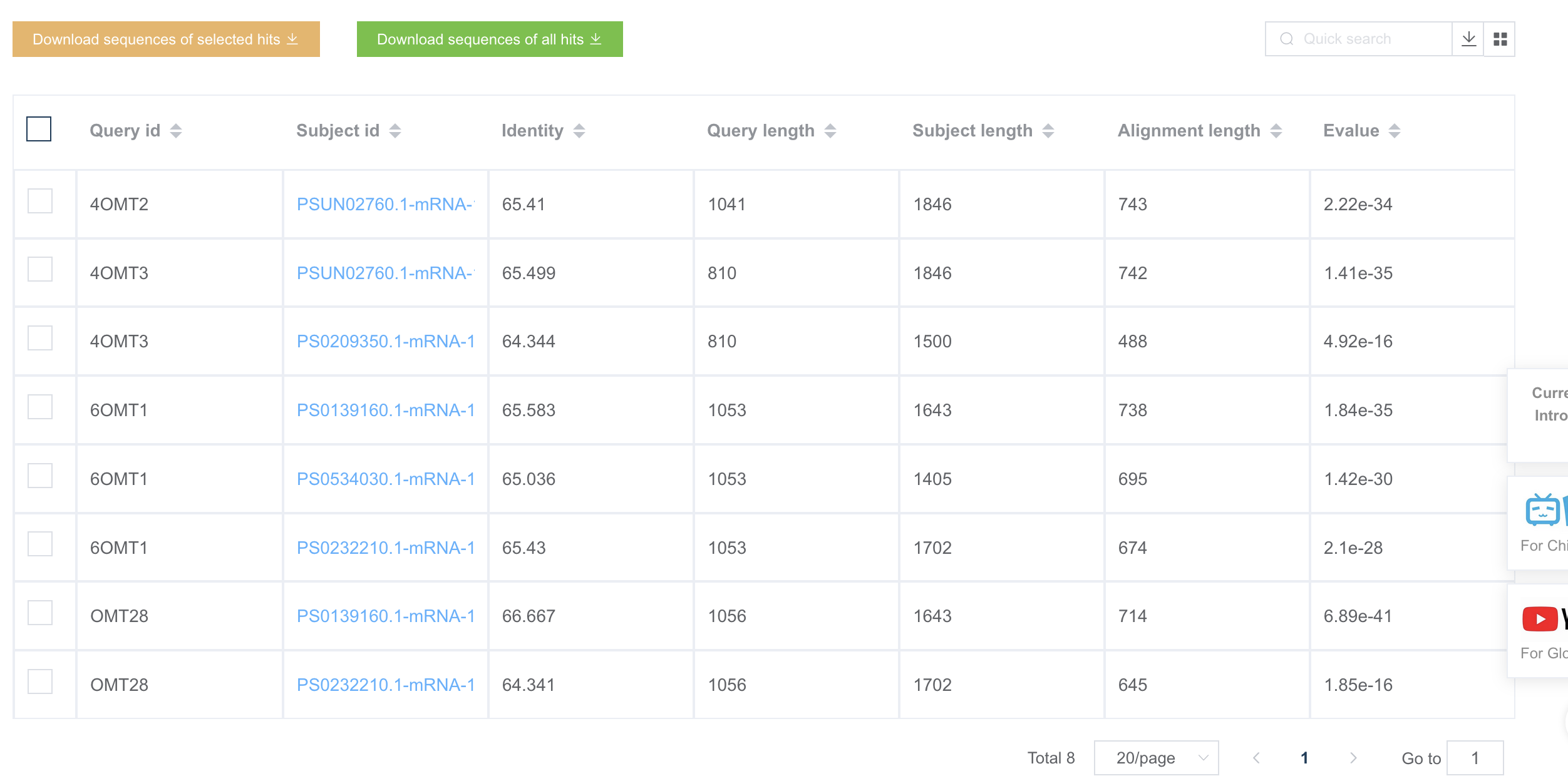
1.8.1 BLAST 参数参考
BLASTN 的匹配得分除以错配罚分 (abs(reward/penalty))的商(比值)越大表示允许的序列直接的匹配度越小。比值为 0.33 等同于序列相似度大于 99%;比值为 0.5 等同于序列相似度大于 95%;比值为 1 等同于序列相似度大于 75%。
It is important to choose reward/penalty values appropriate to the sequences being aligned with the (absolute) reward/penalty ratio increasing for more divergent sequences. A ratio of 0.33 (1/-3) is appropriate for sequences that are about 99% conserved; a ratio of 0.5 (1/-2) is best for sequences that are 95% conserved; a ratio of about one (1/-1) is best for sequences that are 75% conserved
The reward/penalty values are ordered from most to least stringent, with the more stringent values better suited for alignments with high sequence identity.
| reward/penalty | gap costs (open/extend) | default MegaBLAST gap costs (open/extend) |
|---|---|---|
| 1/-5 | 3/3 | 0/5.5 |
| 1/-4 | 1/2, 0/2, 2/1, 1/1 | 0/4.5 |
| 2/-7 | 2/4, 0/4, 4/2, 2/2 | 0/8 |
| 1/-3 | 2/2, 1/2, 0/2, 2/1, 1/1 | 0/3.5 |
| 2/-5 | 2/4, 0/4, 4/2, 2/2 | 0/6 |
| 1/-2 | 2/2, 1/2, 0/2, 3/1, 2/1, 1/1 | 0/2.5 |
| 2/-3 | 4/4, 2/4, 0/4, 3/3, 6/2, 5/2, 4/2, 2/2 | 0/4 |
| 3/-4 | 6/3, 5/3, 4/3, 6/2, 5/2, 4/2 | N/A |
| 4/-5 | 6/5, 5/5, 4/5, 3/5 | N/A |
| 1/-1 | 3/2, 2/2, 1/2, 0/2, 4/1, 3/1, 2/1 | N/A |
| 3/-2 | 5/5 | N/A |
| 5/-4 | 10/6, 8/6 | N/A |
1.10 多序列比对展示
多序列比对是系统进化树构建的前缀,IMP 支持用户直接输入序列或提供基因名字自动提取序列进行多序列比对。
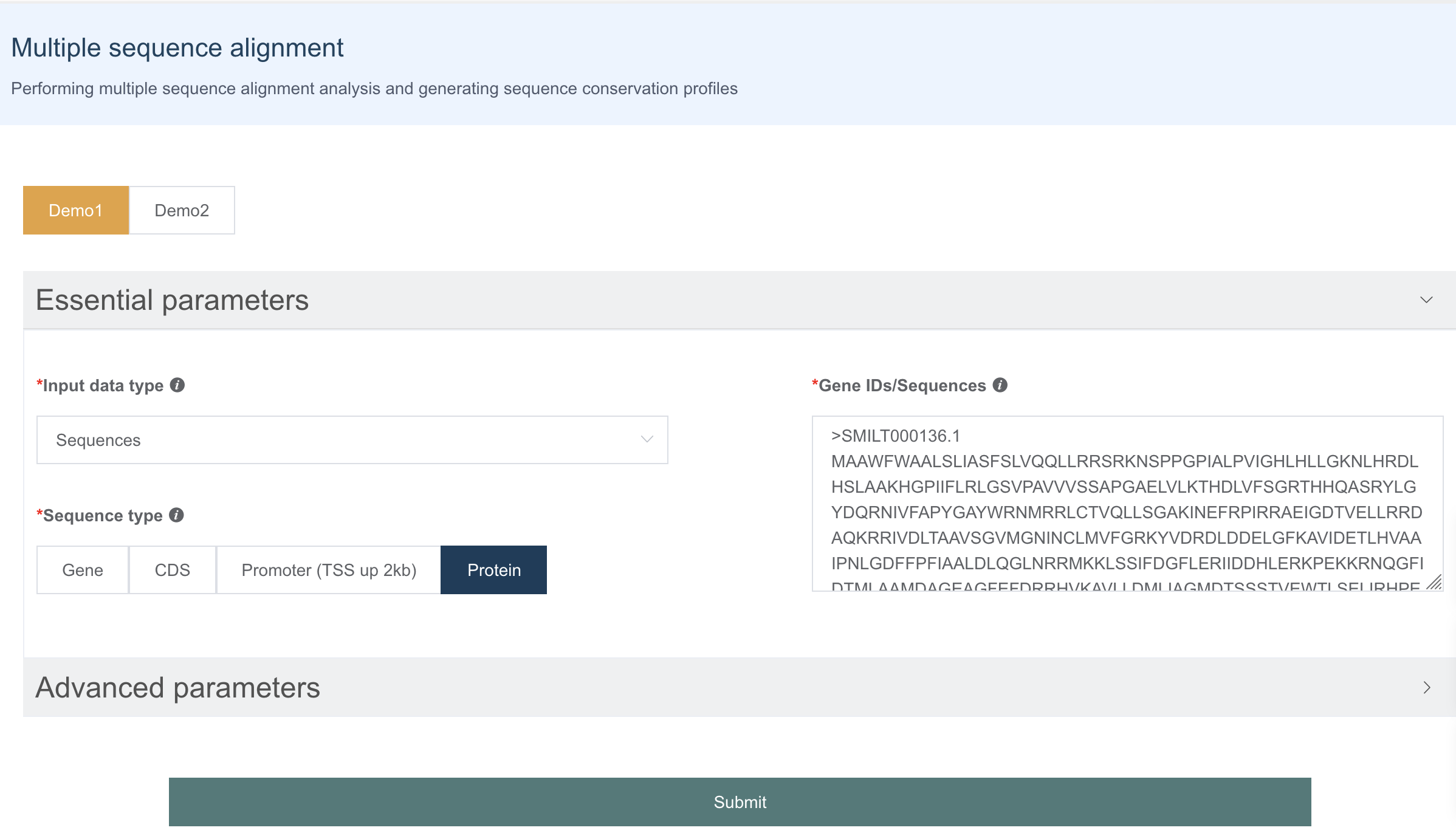
多序列比对展示处,用户可以调整氨基酸或碱基的上色模式、一行展示的序列长度以便获得合适长宽比的可视化图。
1.12 基因簇可视化
Gene map viewer 用于可视化基因组范围的基因分布,查看用户输入的基因是否在染色体区域成簇存在。设计有 2 种展示模式:
- Overlay:显示染色体局部区域的基因分布
- Annotation:显示基因在染色体水平的分布模式
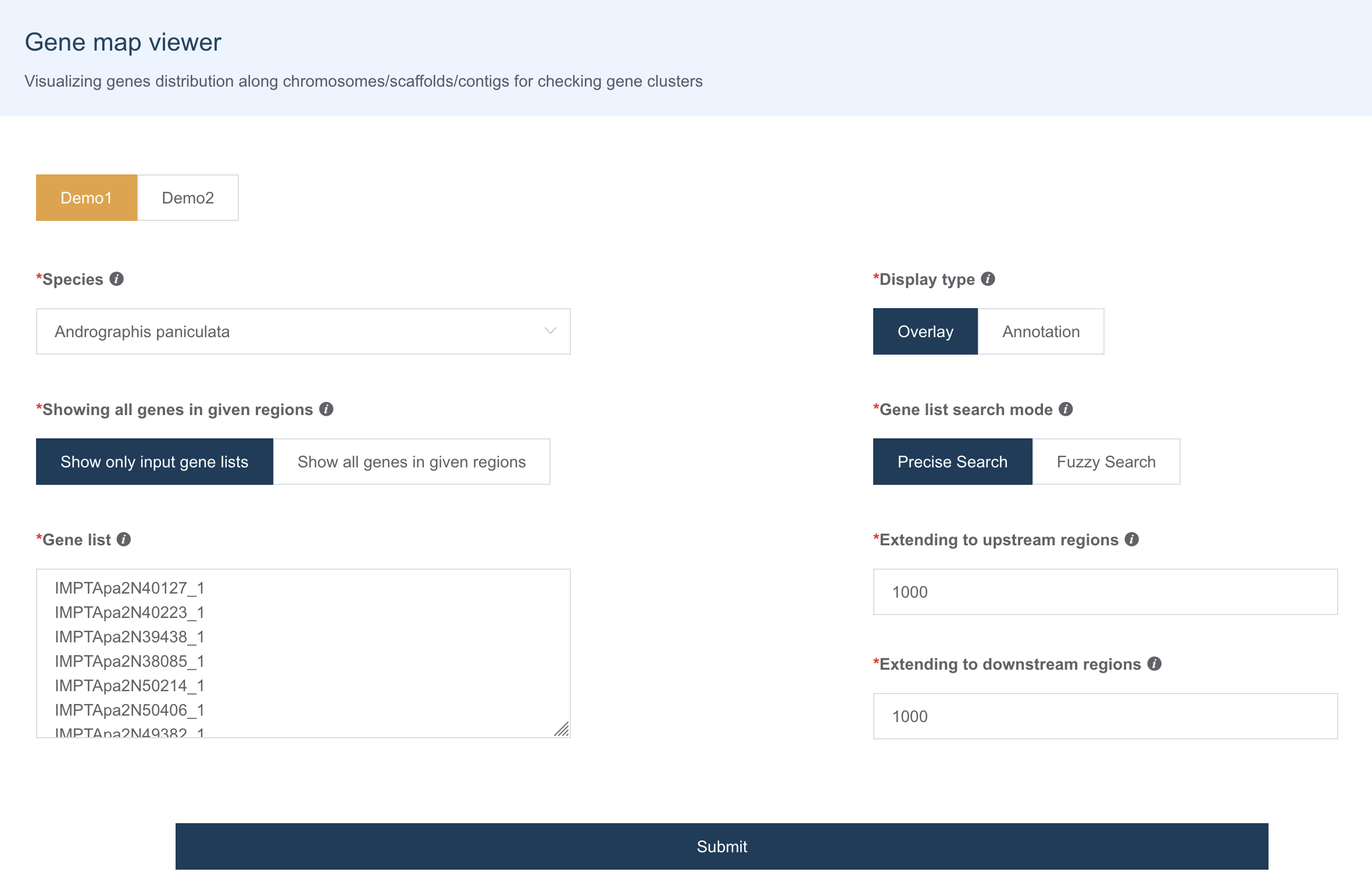
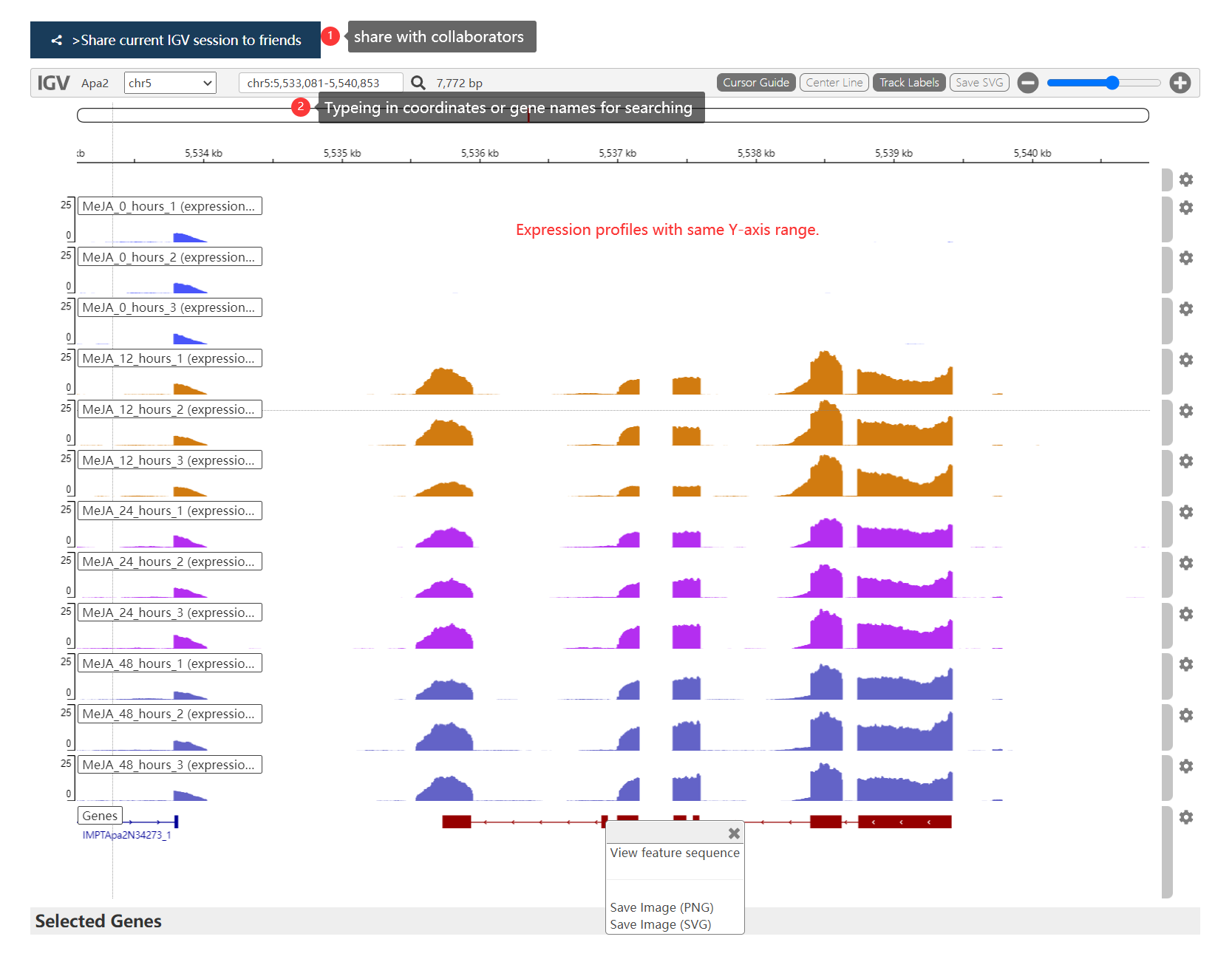
Overlay可视化结果如下, 可以滚动鼠标缩放可视化区域,点击 Gene block 会跳转到 IGV 页面或基因详情页面:

Annotation模式下可视化结果如下, 如果多个基因位置在 0.2M bp内,则合并在一个三角形中展示。
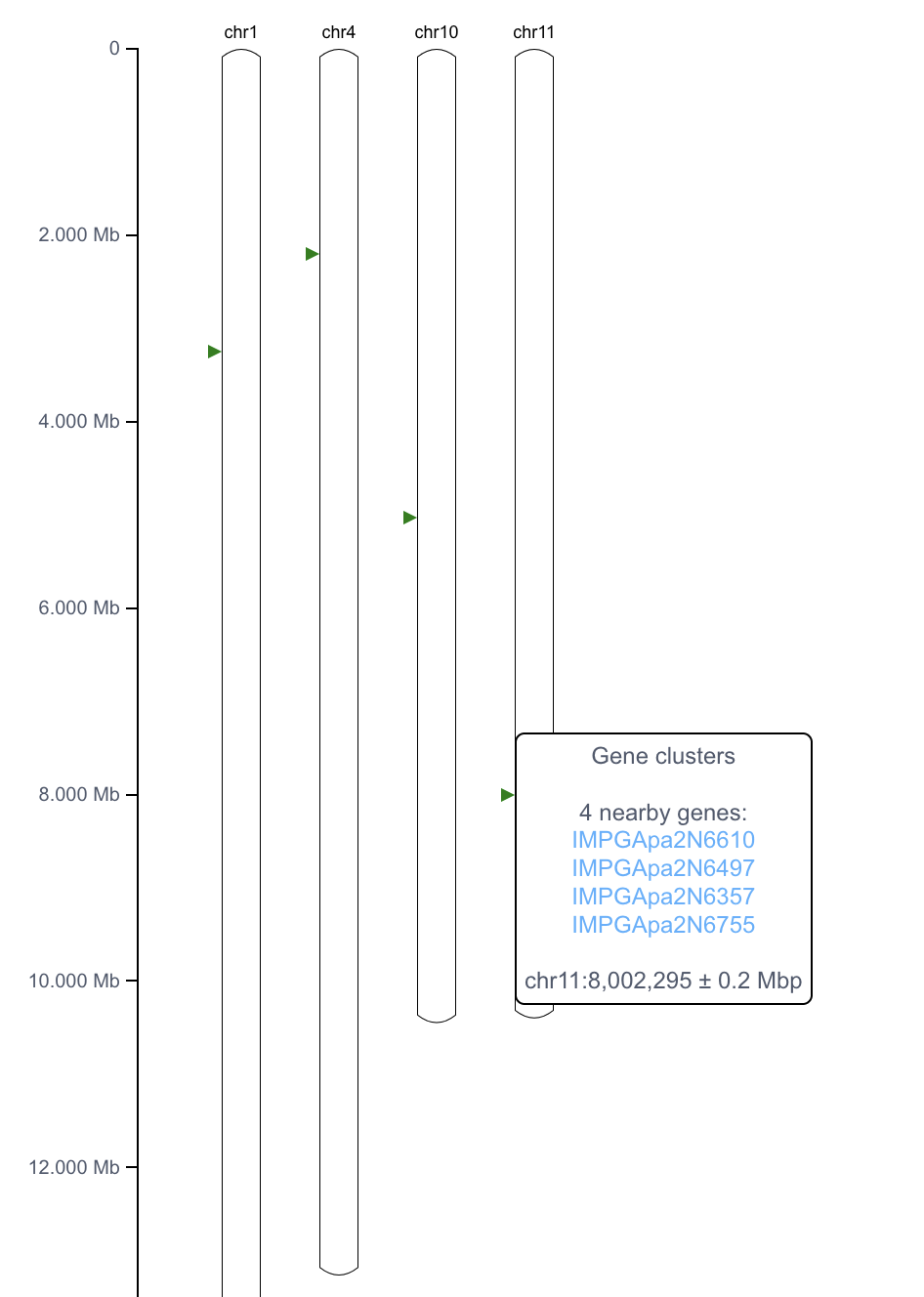
更多基因共线性分析见 https://www.bic.ac.cn/SynColV。
1.13 在线实验设计和差异基因分析
本部分基于Reads-count矩阵采用limma-voom 进行差异基因的鉴定,然后对筛选出的差异基因进行GO/KEGG富集分析.
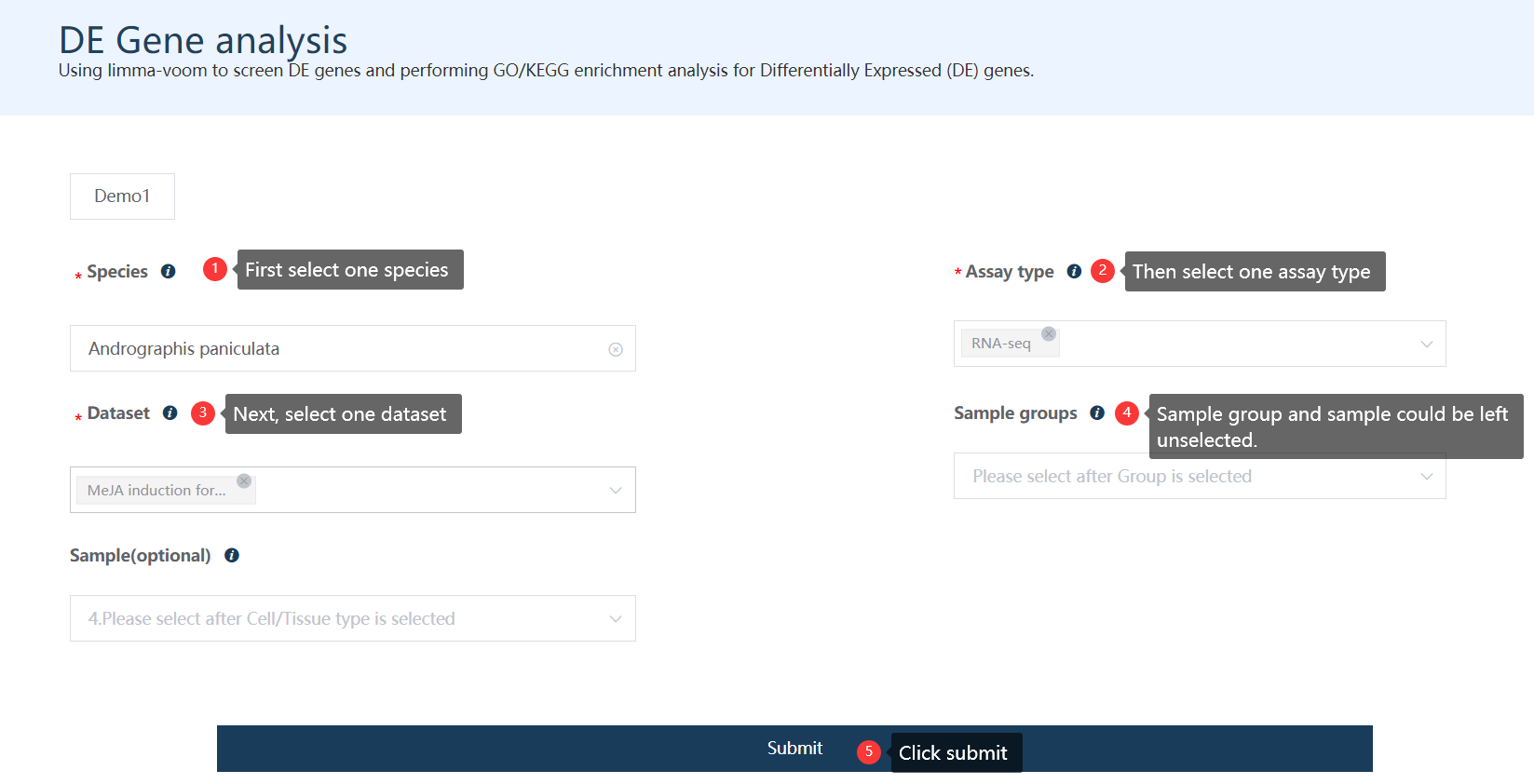
该功能涉及多个分析步骤,每个步骤页面结构差不多,下图是对于表单部分的解释。

Figure 1.1: A screenshot of the form structure.
1.13.2 第二步:样品相关性评估和过滤异常样品
针对选中的样品,提取其表达矩阵,并采用DESeq2 类似的方式计算量化因子获得标准化后的数据矩阵,然后绘制样品相关性热图和 PCA 分析。
下图中的左右穿梭框显示了系统自动鉴定出的异常样品和通过检测的样品,用户也可以根据下面的可视化结果自行调整或筛选样品。
![]()
下面展示的是样品聚类热图和 PCA 分析的结果图,二者都是交互式图谱。
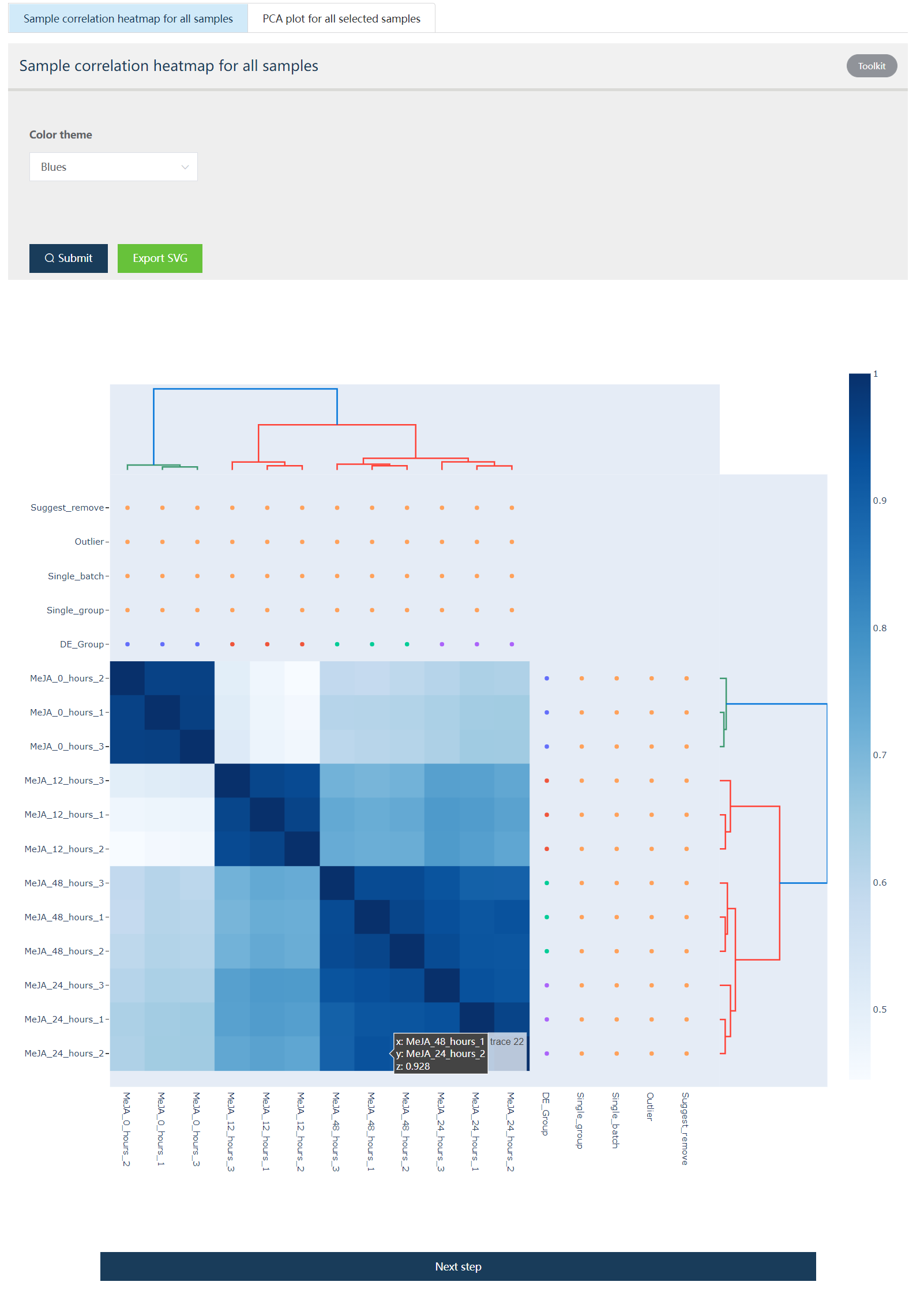
Figure 1.2: 12个样品的表达相关性热图展示。行列注释中的DE_Group: 用户选择数据的生物分组信息。Single_group: 检查是不是有某个组只有 1 个生物学重。Single_batch: 检查某个批次的数据是不是只有 1 个样本。Outlier: 标识系统鉴定出的异常样品。Suggest_remove: 建议移除的样品。
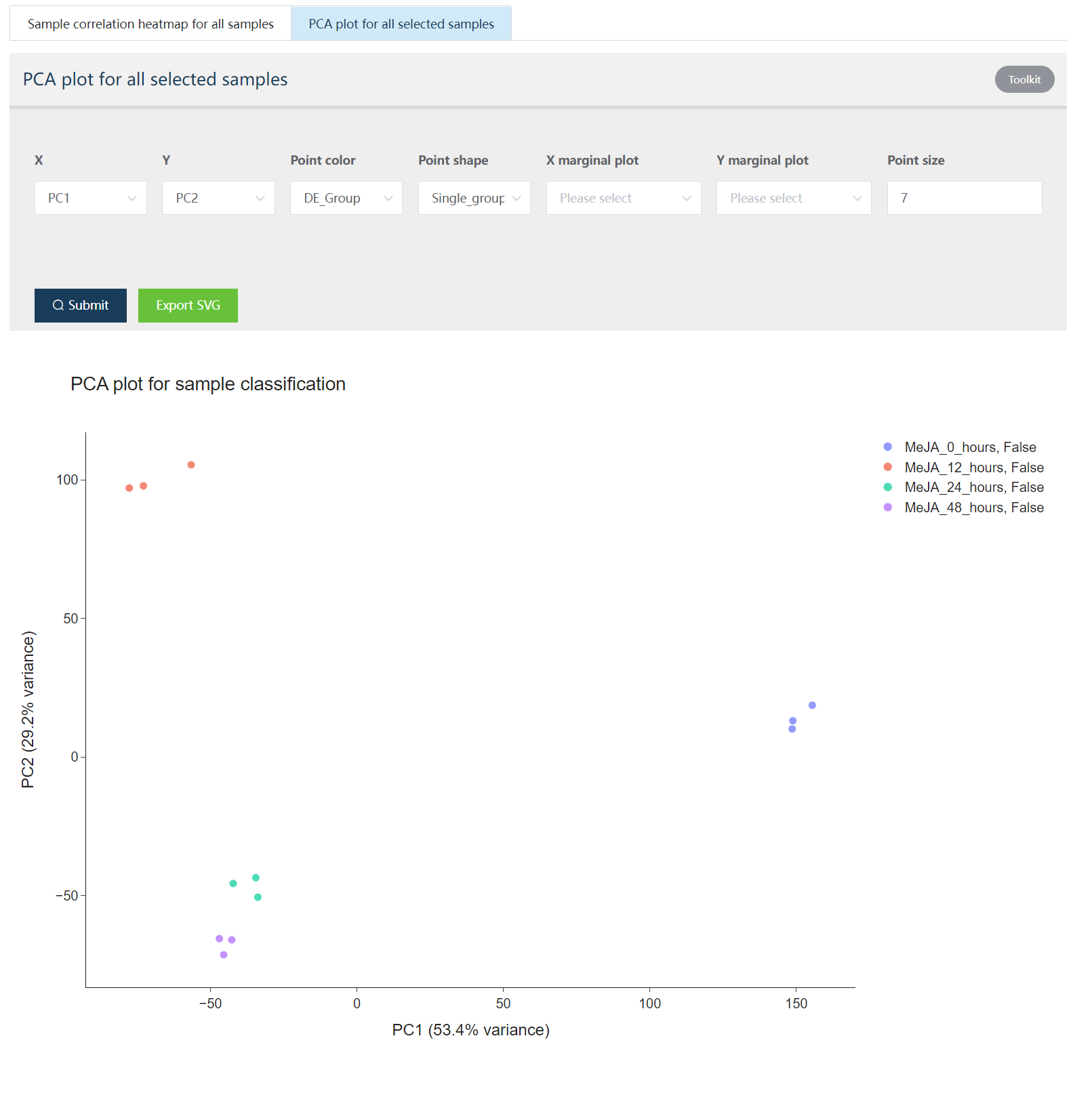
Figure 1.3: 可视化样品在主成分分析获得的第 1 和 2 组成分构成的空间中的分布. Toolkit 部分用户可以选择其它主成分进行展示,也可以调整点的颜色、大小、形状和绘制数据的分布模式。
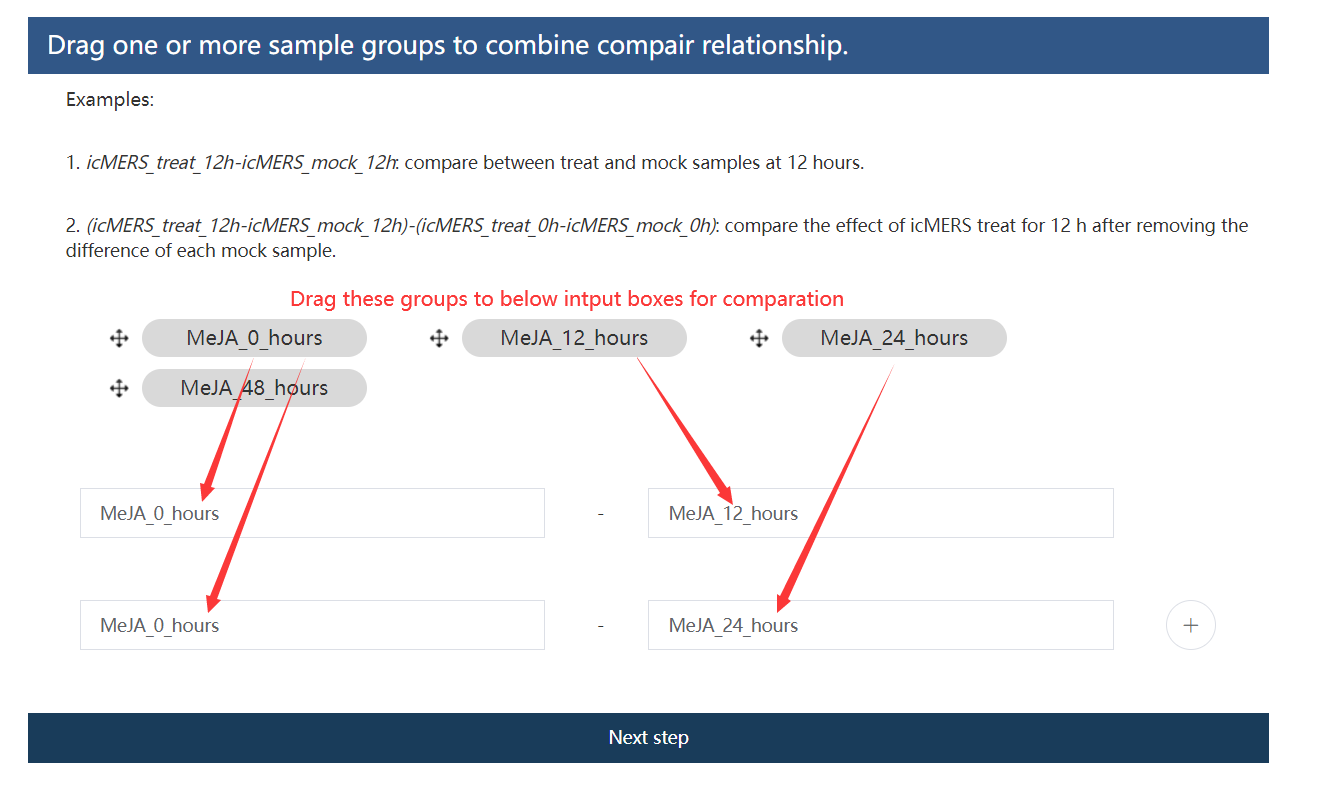
1.13.4 第四步:设置差异基因过滤阈值
计算出的 FDR 值低于用户指定的值且表达变化倍数高于用户指定的值得基因定义为差异基因。
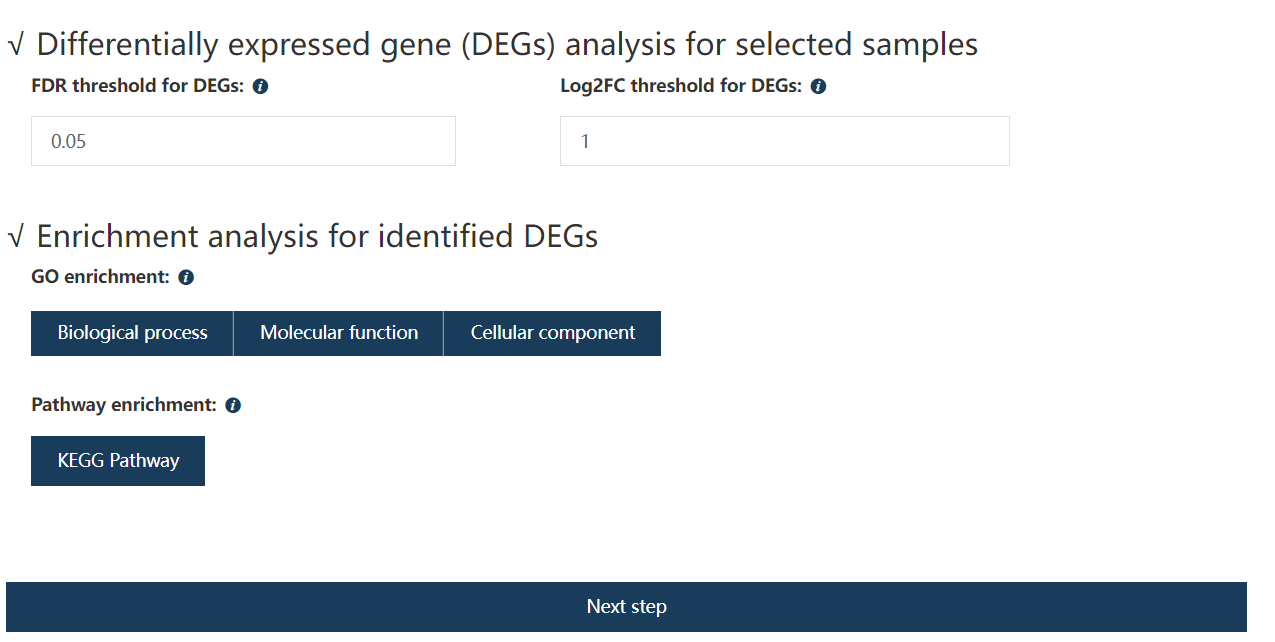
Figure 1.5: Set the FDR and absolute Log2FC for screening DE genes.
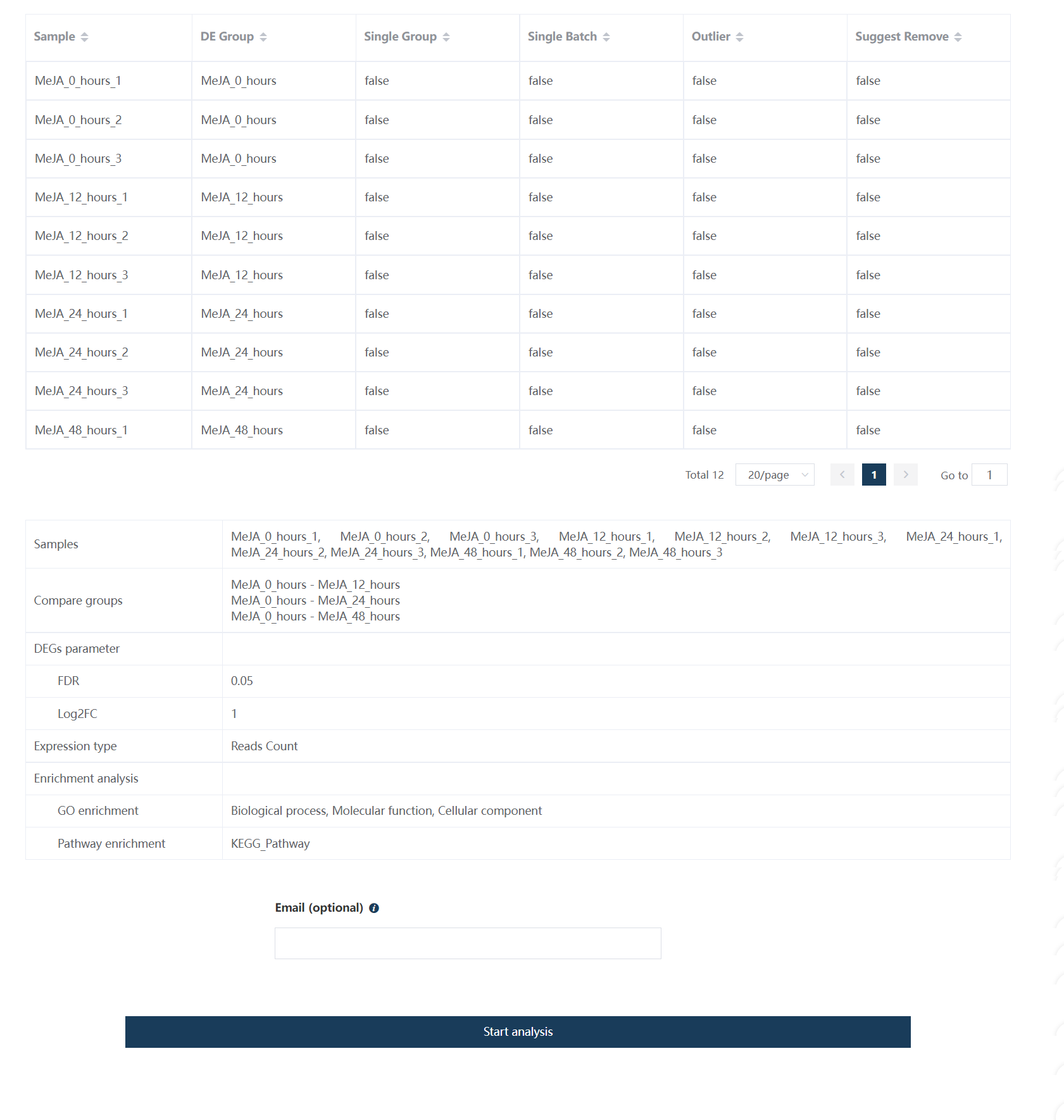
1.13.6 第六步:差异基因分析结果报告
差异基因分析结果报告包含样品信息、样品相关性热图、PCA 分析、差异基因热图、差异基因火山图、功能富集分析结果等。每一部分结果图都可以做进一步定制,也可以导出数据,放到一款高颜值免费在线SCI绘图工具ImageGP做更多可视化分析。

Figure 1.7: 目录展示结果报告整体内容,各个部分可点击直接跳转。
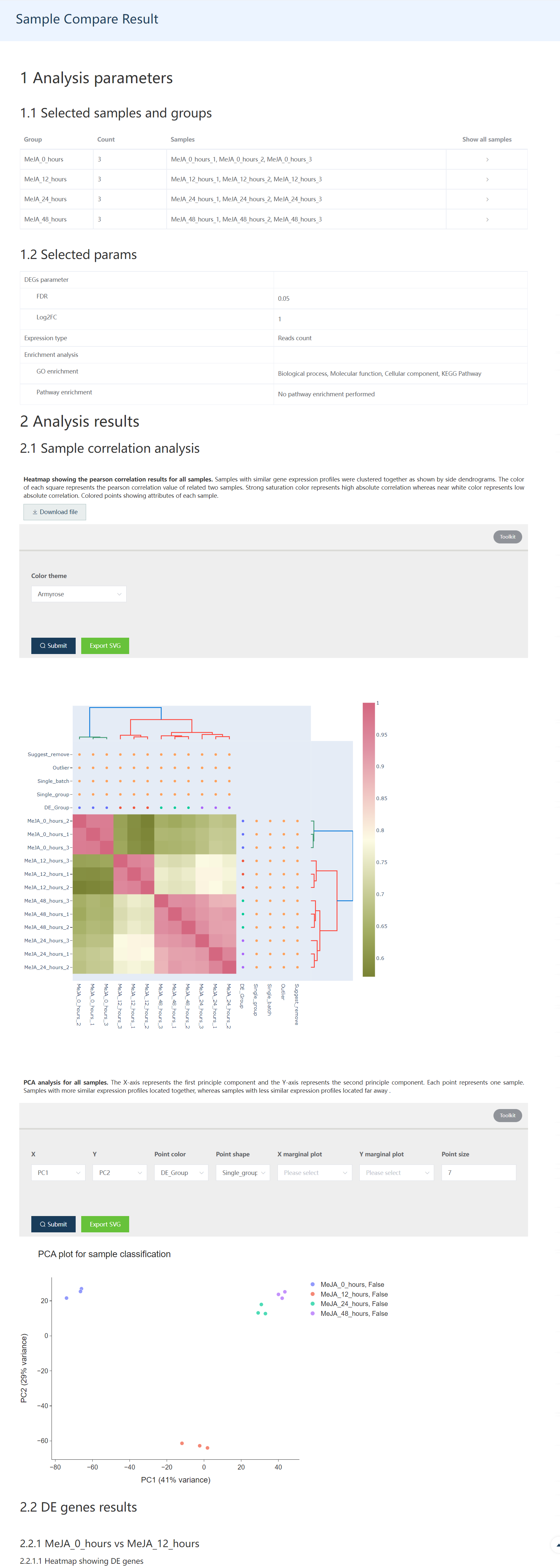
Figure 1.8: The first part of the DE gene result report includes whole sample information, and sample correlation information.
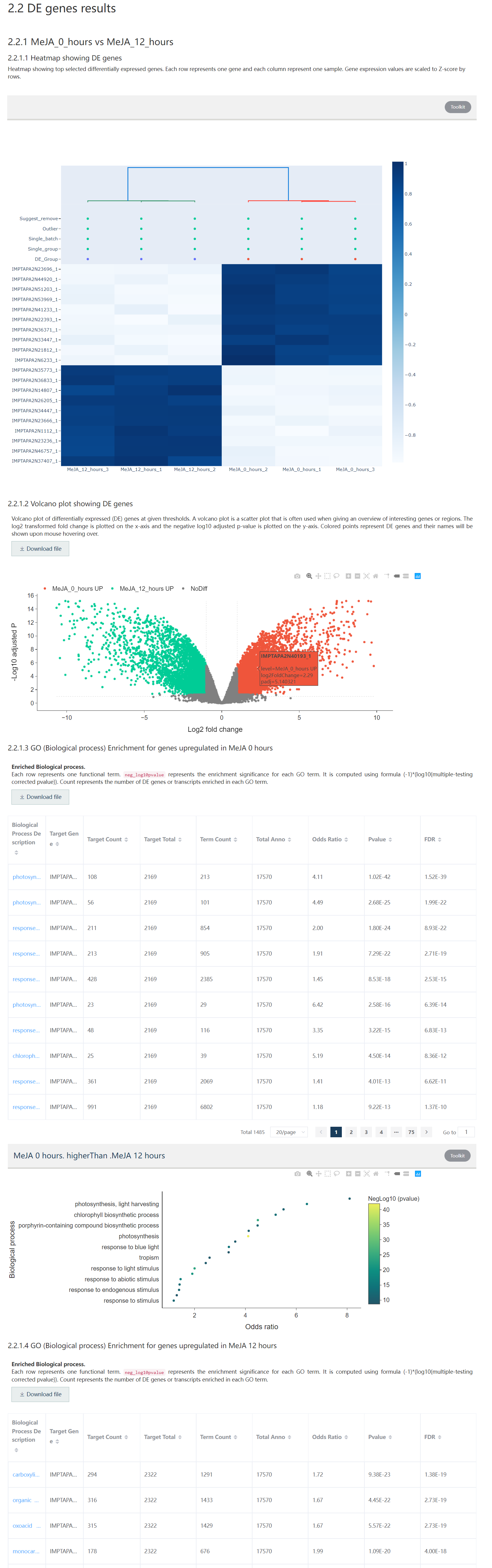
Figure 1.9: The second part of the DE gene result report includes DE gene heatmap, volcano plot, and functional enrichment result.
整个结果也可以导出为 PDF 格式:当所有结果完成加载后,按Ctrl + p会启动Printer to PDF或打印到 PDF 功能,点击确认后即可输出 PDF。